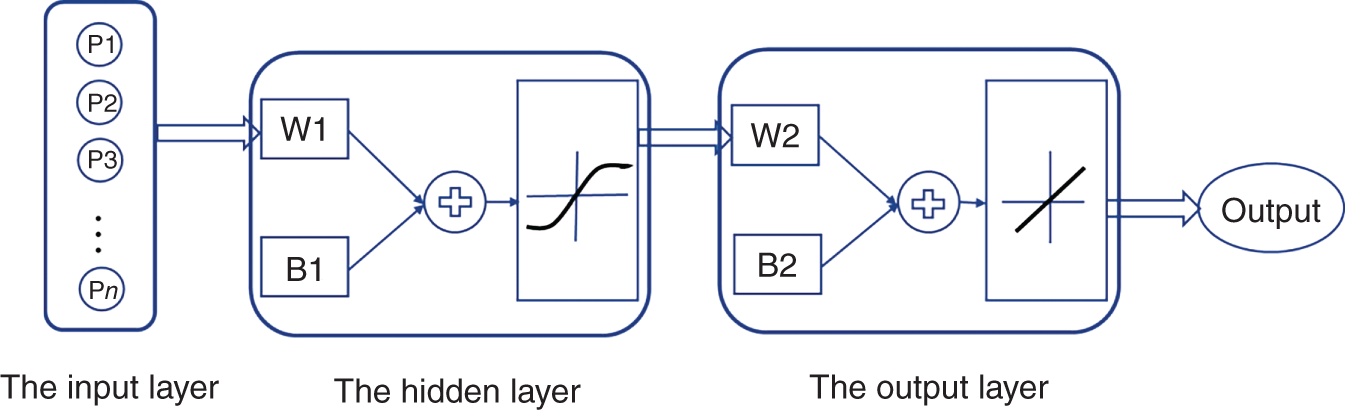
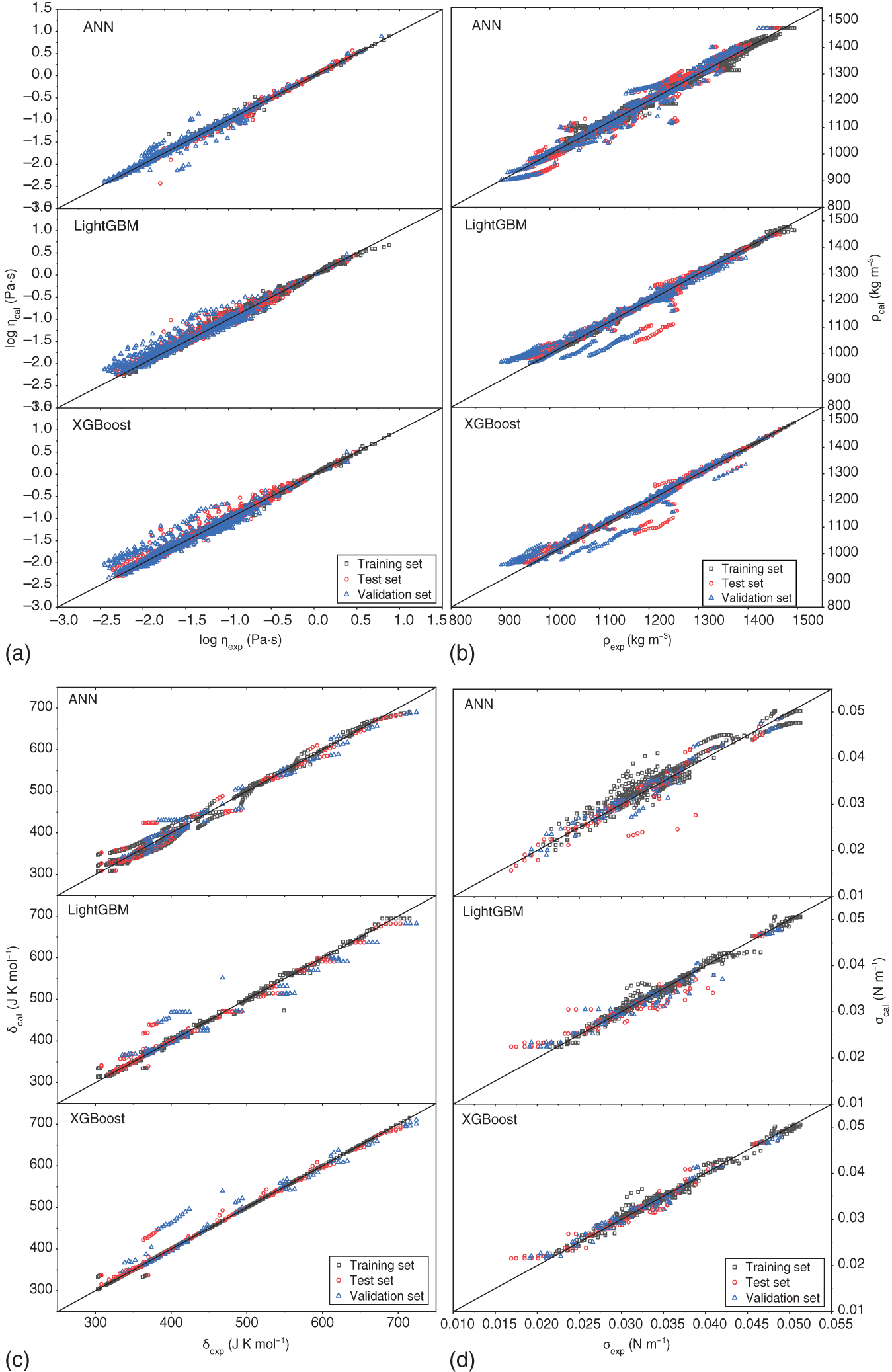
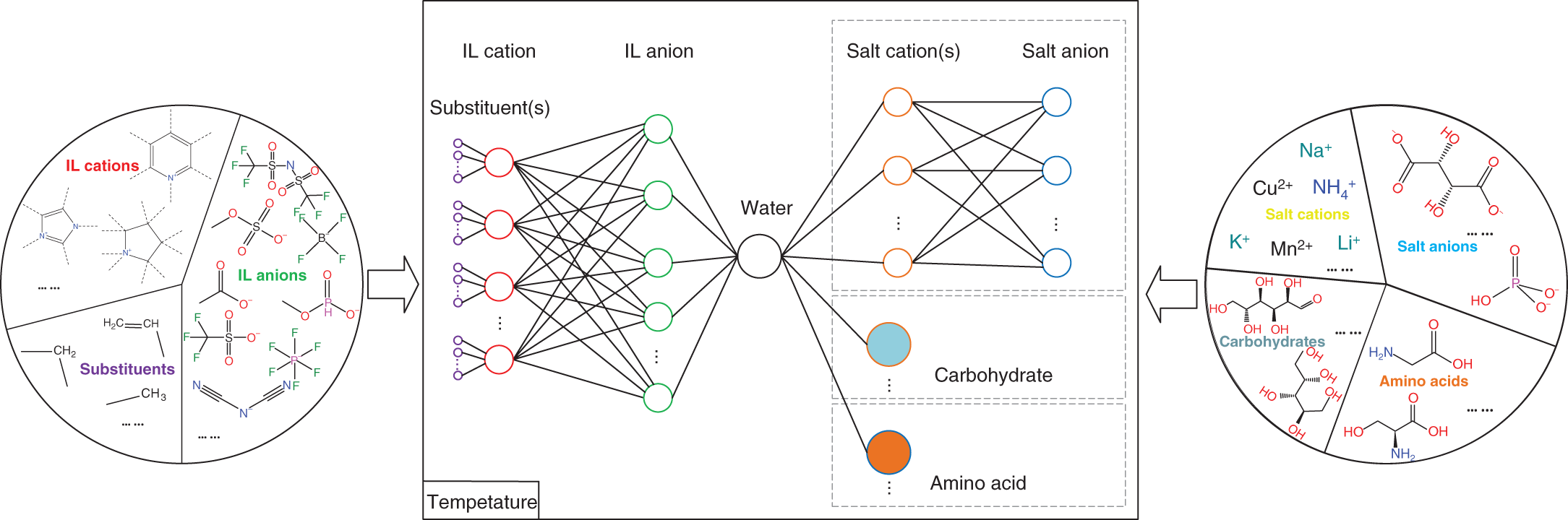
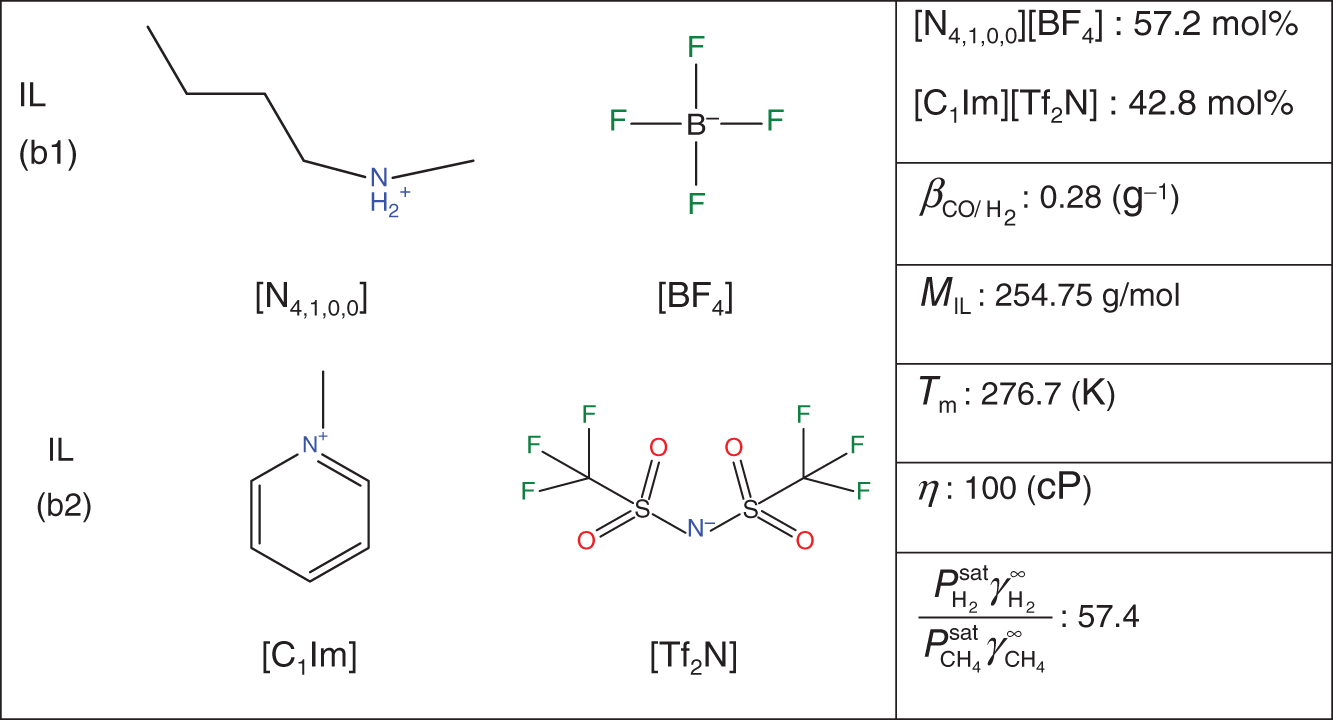
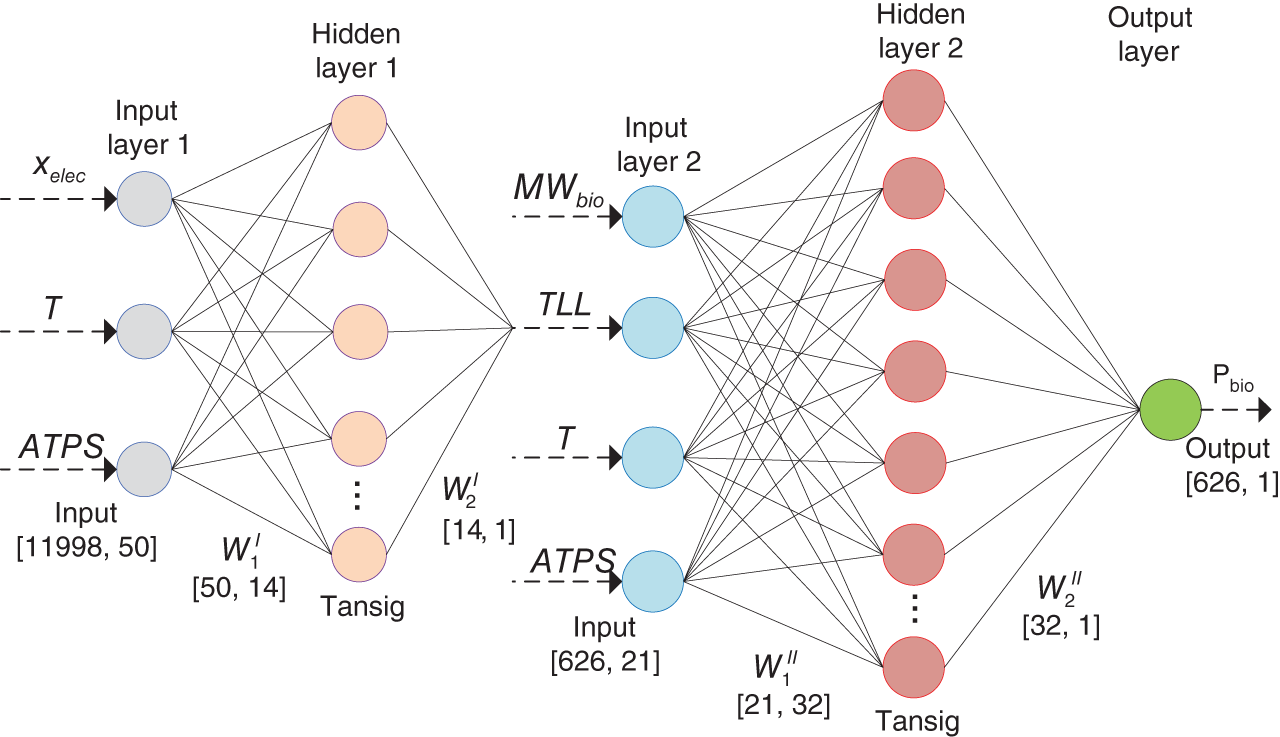
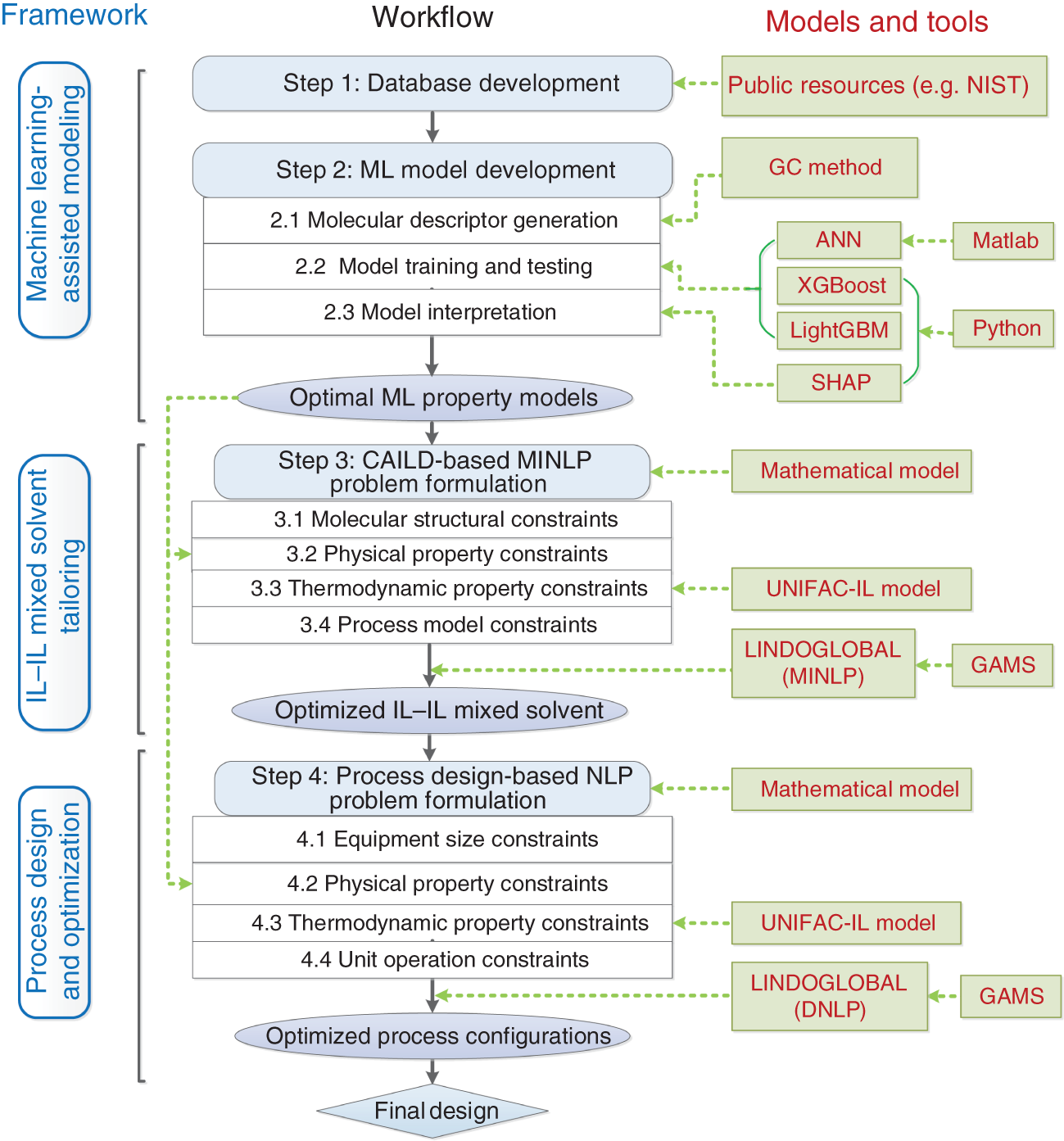
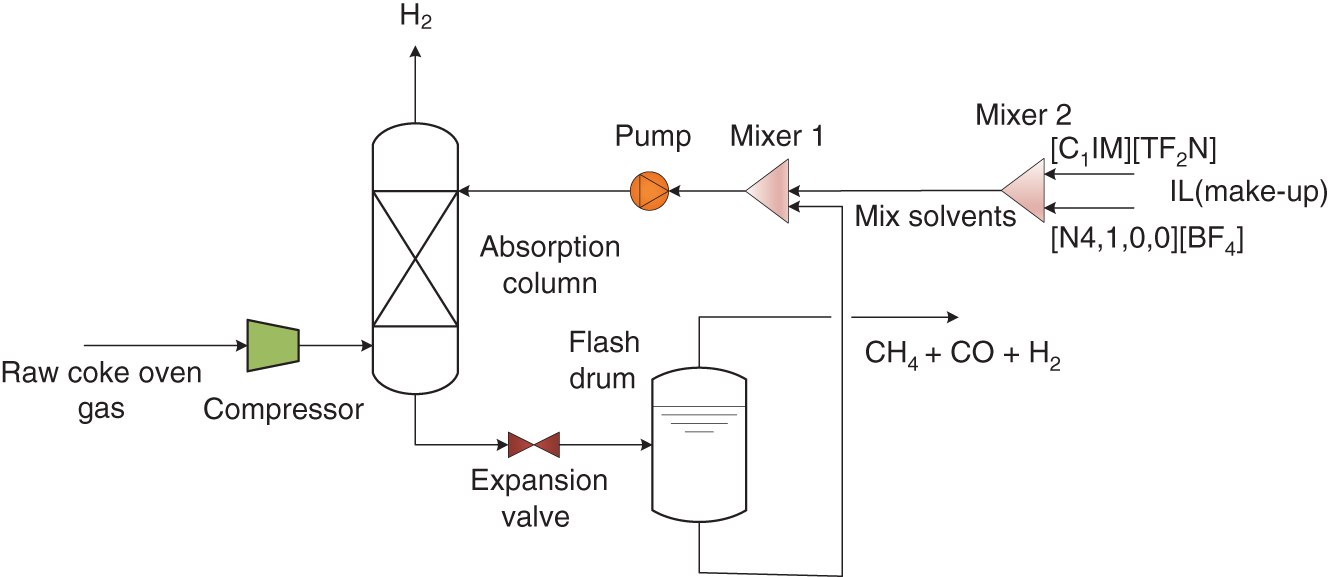
Yuqiu Chen University of Delaware, Department of Chemical and Biomolecular Engineering, 150 Academy Street, Newark, DE 19716, USA The accurate prediction of physical properties is critical for the successful application of both chemicals and materials across various industries. Property modeling using artificial intelligence (AI) has emerged as a powerful and efficient approach in various scientific and engineering disciplines [1]. This methodology leverages advanced algorithms and machine learning (ML) techniques to predict and model the physical and chemical properties of materials, compounds, or systems [2]. One of the key advantages of employing AI in property modeling is its ability to handle complex relationships and patterns within large datasets, leading to more accurate predictions. ML models, such as neural networks and support vector machines (SVMs), can be trained on diverse datasets, enabling them to capture intricate correlations that traditional modeling methods might overlook [3]. In the field of materials science, AI-driven property modeling has proven valuable for predicting properties like conductivity, thermal conductivity, and mechanical strength [4]. Similarly, in chemistry, AI has been applied to forecast molecular properties, solubility, and reaction outcomes [5]. Given the broad scope of AI techniques, the author has chosen to focus on their application in property modeling, solvent tailoring, and process design for systems involving ionic liquids (ILs) and aqueous two-phase systems (ATPSs). ILs are innovative fluids that have garnered significant attention from both academia and industries [6]. Over the past few decades, extensive research has been conducted on the applications of ILs in various fields such as electrochemistry [7], synthetic materials [2, 8], and pharmaceutical manufacturing [9, 10]. In these areas, ILs serve different roles, including as extractants/absorbents in separations, media and/or catalysts in lignocellulosic biomass pretreatment, and functional materials in batteries. With the growing interest in ILs within industrial settings, it is crucial to deeply understand their property behaviors for effective product and process design. For example, viscosity is a crucial transport property that plays a significant role in fluid flow analysis, process optimization, and material characterization [11]. Understanding and controlling viscosity enable engineers, researchers, and industries to design efficient systems, optimize processes, develop high-quality products, and ensure the desired performance and functionality of materials and fluids in a wide range of applications. Another example is that heat capacity is crucial for thermodynamic calculations, energy balance analyses, temperature control systems, material selection, process optimization, and fundamental research. It plays a crucial role in various scientific, engineering, and industrial applications, enabling the efficient and effective design, operation, and optimization of processes involving heat transfer and energy conversion [12]. Consequently, the characterization of IL properties and the establishment of structure–property relationships for ILs and IL-based mixtures are equally important in investigating their applications. ATPSs, also known as aqueous biphasic systems (ABSs), are formed when two or more water-soluble components, such as polymers, salts, ILs, alkaline, and alcohols, are mixed in water at appropriate concentrations and temperatures [13]. To date, various combinations of phase-forming agents (e.g. polymer–salt/alkaline, polymer–polymer, ionic liquid–salt, and alcohol–salt) have been proposed for the creation of ATPSs [14]. Many ATPSs and the combination of these two-phase systems with other techniques such as microfluidic apparatus [15], have exhibited great technical and economic advantages in biotechnological applications [16]. Due to the high water content in both phases, ATPSs can provide a biocompatible and nondenaturing environment for cells, proteins, and other biomolecules. Meanwhile, ATPSs generally present less damage to the extracted biomolecules as they allow rapid phase separation and compound partition, leading to much lower interfacial stress than that of organic-water solvent systems. In addition, ATPSs can offer high recovery percentages and high purity of biomolecules in a one-step process. Besides these, ATPSs show characteristics of high tailored space, and they are also easy to scale up [17]. A good understanding of the mechanism that can guide the phase formation of ATPS is obviously of great importance for enhancing the opportunity for ATPS adoption in the industry. Meanwhile, the ability to provide reliable predictions on the partition of biomolecules in ATPS is also essential, given the fact that it would largely reduce the time and cost to find high-performance ATPS for biomolecules. Therefore, a systematic modeling study on the phase equilibria behavior of ATPS and the partition of biomolecules in ATPS is highly desirable for the transition of ATPS separation technique from pure academic focus to industrial implementation. Due to the high complexity of ATPS and IL-involved systems, empirical correlations and theory-driven models cannot simultaneously provide reliable physical and thermodynamic predictions. In this respect, AI techniques such as ML algorithms are potential alternatives to model thermodynamic and transport properties of complex systems [18] such as IL-based ATPS and ionic liquid–water mixtures. The advantages of using AI techniques for modeling complex systems include the following: (i) AI techniques, particularly ML, can handle complex patterns and nonlinear relationships in data, making them well-suited for modeling intricate chemical properties. (ii) AI models can provide accurate predictions for various chemical properties. This accuracy is crucial in industries such as pharmaceuticals, materials science, and environmental research, where precise chemical property information is essential. (iii) Chemical data often involves a high number of dimensions, with numerous variables influencing the properties of interest. AI algorithms, especially those used in deep learning, excel at extracting relevant features from high-dimensional datasets. (iv) AI techniques facilitate the integration of diverse data types, including experimental results, literature data, and computational simulations. This comprehensive approach enhances the accuracy and reliability of chemical property models. (v) The increasing availability of large datasets in chemistry, including databases of chemical compounds and their properties, aligns well with AI’s ability to handle big data. AI can uncover meaningful patterns and correlations within massive datasets. (vi) AI techniques can be applied across a range of chemical domains from organic chemistry to materials science. Their adaptability makes them versatile tools for modeling diverse chemical properties. Recently, ML algorithms, including multiple linear regression (MLR), k-nearest neighbor (KNN), decision tree (DT), random forest (RF), gradient boosting (GB), artificial neural network (ANN), multilayer perceptron (MLP), SVM, XGBoost, and lightweight gradient-boosting machine (LightGBM), have been employed as property prediction tools for complex systems. The advantages and disadvantages of these ML algorithms are described in the following text: MLR is a supervised ML algorithm used for predicting the numerical value of a dependent variable based on the values of two or more independent variables. It is a foundational and widely used technique, especially when dealing with datasets where the outcome is influenced by multiple factors. MLR serves as the basis for more advanced regression techniques in ML. The KNN algorithm is a versatile and intuitive ML algorithm used for both classification and regression tasks. KNN is a valuable tool in various applications, including classification problems in image recognition, recommendation systems, and anomaly detection. KNN is easy to understand and implement, making it a good choice for initial exploration of a dataset. It does not involve a training phase in the traditional sense, and the model is trained during the prediction phase. KNN can be applied to various types of datasets and is effective in capturing complex patterns. However, KNN may struggle with imbalanced datasets, where one class significantly outnumbers the others. In addition, calculating distances between data points for large datasets can be computationally intensive. Nonetheless, its simplicity and ease of implementation make it a valuable tool, especially in cases where interpretability and explainability are important. Decision tree is a versatile and widely used ML algorithm that can be applied to both classification and regression tasks. It is a supervised learning algorithm that recursively partitions the data into subsets based on the features, ultimately leading to a decision or prediction. Decision trees are easy to interpret and visualize. The decision-making process is transparent, allowing users to understand how predictions are made. Decision trees can be prone to overfitting, especially if the tree is deep and complex. Regularization techniques like pruning or using a minimum number of samples per leaf can help mitigate this. Decision trees serve as the foundation for more advanced ensemble methods like RF and GB. RF is an ensemble learning method in ML that operates by constructing a multitude of decision trees during training and outputting the class that is the mode of the classes (classification) or mean prediction (regression) of the individual trees. RF often provides higher accuracy compared to individual decision trees, especially for complex datasets. The ensemble approach and feature randomization help mitigate overfitting, making RF more robust. RF can be applied to both classification and regression tasks and can handle missing values in the dataset without the need for imputation. However, RFs are less interpretable compared to individual decision trees, as the combined effect of multiple trees can be complex. In addition, RF can be memory intensive, especially with a large number of trees or features. Nonetheless, RF is widely used in practice and is considered a powerful and versatile algorithm. It is effective for a variety of tasks, including classification, regression, and feature selection. Its robustness and high accuracy make it a popular choice in the ML community. GB is a powerful ensemble learning technique used for both classification and regression tasks. It is a boosting algorithm that combines the predictions of multiple weak learners (typically decision trees) sequentially, with each tree correcting the errors of the previous ones. GB can capture complex, nonlinear relationships in the data and often achieves high accuracy, making it one of the state-of-the-art algorithms for many ML tasks. However, GB can be prone to overfitting, especially if the number of trees is too large, and it can be computationally intensive when training a large number of trees. Nonetheless, GB is widely used in practice and has proven effective in various ML tasks. It is particularly popular in Kaggle competitions and real-world applications where accurate predictions are essential. ANN is a computational model inspired by the structure and function of the human brain. It consists of interconnected nodes, called artificial neurons or “neurons,” organized into layers. Each neuron takes input, performs a computation, and produces an output that is passed on to other neurons. The advantages of ANN in modeling studies can be summarized as follows: (i) ANNs can learn organically, meaning their outputs are not limited solely by inputs, and they have the ability to generalize from their inputs. (ii) Nonlinear systems can find shortcuts to reach computationally expensive solutions. (iii) ANNs have high fault tolerance potential. (iv) ANNs can perform tasks beyond routing around nonoperational parts of the network. The advantages of ANNs are particularly evident when a large experimental dataset with a wide range of variables is available. MLP is a type of ANN and a fundamental ML algorithm. It falls under the category of supervised learning and is widely used for both classification and regression tasks. Figure 1.1 presents a structure of the three-layer ANN with an input vector size of 46 × 1. SVM is a supervised ML algorithm used for both classification and regression tasks. The primary goal of SVM is to find a hyperplane that best separates the data into different classes in a high-dimensional space. For a binary classification task, SVM aims to find a hyperplane that separates data points of one class from another. For regression tasks, SVM seeks to find a hyperplane that best fits the data. In a two-dimensional space, a hyperplane is a line. In higher dimensions, it becomes a hyperplane. Support vectors are the data points that are closest to the decision boundary (hyperplane). SVM can use a kernel function to map the input features into a higher-dimensional space. Common kernel functions include linear, polynomial, radial basis function (RBF), and sigmoid. The optimization objective is to maximize the margin between classes. SVM solves a quadratic optimization problem to find the optimal hyperplane and support vectors. SVM is effective in high-dimensional spaces, making it suitable for tasks with many features. It is widely used in various applications, including image recognition, text classification, and bioinformatics. Proper tuning of parameters, especially the choice of the kernel and regularization parameters, is crucial for achieving optimal performance with SVM. Figure 1.1 Structure of a three-layer ANN with an input vector size of n × 1. Source: [19]/with permission of Elsevier. XGBoost is a parallel regression tree model based on the boosting technique, where boosting refers to obtaining the final classifier by weighting the sum of existing weak classifiers. The XGBoost model is an improvement over the Gradient Boosted Decision Tree (GBDT) model. Compared to GBDT, XGBoost significantly enhances the speed of model training calculations and improves prediction and classification accuracy, making it an upgraded version of the GBDT algorithm. To prevent model overfitting and improve generalization ability, XGBoost incorporates regularization terms into the loss function of the GBDT model. The traditional GBDT loss function adopts a first-order Taylor expansion and uses the negative gradient value to replace the residual for fitting. In XGBoost, a second-order Taylor expansion is added to the loss function, capturing the second derivative to gather gradient direction information and improve model accuracy. Furthermore, XGBoost employs block-wise and sorted feature processing, enabling parallelization for finding the best-split points and thereby enhancing computation speed. LightGBM is a GBDT algorithm framework renowned for its rapid training, minimal memory requirements, support for efficient parallel training, enhanced accuracy, and the ability to process extensive datasets swiftly. While XGBoost is a widely recognized GBDT tool, it grapples with significant memory usage. To tackle this challenge, Microsoft developed LightGBM as an optimization of the conventional GBDT algorithm. LightGBM distinguishes itself from XGBoost by implementing the histogram algorithm instead of the presorted algorithm. This choice results in decreased memory consumption and simplifies data separation. However, it is worth noting that the histogram algorithm may not be as time efficient as the presorted algorithm when processing sparse data. LightGBM incorporates two novel techniques, Exclusive Feature Bundling (EFB) and Gradient-based One-Side Sampling, into the histogram-based GBDT algorithm. EFB enables the fusion and bundling of certain features, reducing the feature dimension without sacrificing accuracy. Additionally, LightGBM employs a leaf-wise strategy with a capped maximum depth, which serves to prevent overfitting without compromising computational efficiency. To date, integrating ML algorithms into property modeling has been widely studied for different systems, including ATPS and IL-involved systems [20]. Yusuf et al. (2020) [21] reviewed the application of AI-based predictive methods in IL studies. Koutsoukos et al. (2021) [22] conducted a critical review of the use of ML algorithms as property prediction tools for the viscosity, density, melting point, and toxicity of pure ILs as well as the solubility of acid gases (H2S, CO2) in ILs. Therefore, the discussion in this work focuses on studies not covered in Yusuf’s and Koutsoukos’s review. For pure ILs, Baskin et al. (2022) [20] combined three traditional ML algorithms and neural networks with seven different architectures with five types of molecular representations (in the form of either numerical molecular descriptors or simplified molecular input line entry system (SMILES) text strings) to construct quantitative structure-property relationship (QSPR) models for predicting six important physical properties of ILs: density, electrical conductivity, melting point, refractive index, surface tension, and viscosity. The results showed that (i) nonlinear ML algorithms perform much better than linear ones, (ii) neural networks perform better than traditional ML methods, and (iii) transformers that are actively used in natural language processing (NLP) perform better than other types of neural networks due to the advanced ability to analyze chemical structures of ILs encoded into SMILES text strings. Dhakal and Shah (2021) [23] applied SVM and ANN to correlate ionic conductivity of imidazolium-based ILs. Both models were shown to successfully capture the entire range of ionic conductivity spanning 6 orders of magnitude over a temperature range of 275–475 K with relatively low statistical uncertainty, and the ANN-based model presented slightly better performance. The performance of the ANN algorithm in the modeling study of the conductivity of ILs was also validated by Datta et al. (2022) [24]. Dhakal and Shah (2022) [25] further applied three ML algorithms (MLR, RF, and XGBoost) to conduct models for the conductivity prediction of ILs, and XGBoost performed the best. Meanwhile, Karakasidis et al. (2022) [26] incorporated six different numerical ML algorithms, namely MLR, KNN, DT, RF, gradient boosting regressor (GBR), and MLP for electrical conductivity prediction. Results showed that all ML algorithms performed well on their predictions, while the best fit was obtained for the GBR algorithm. Yalcin et al. (2019) [27] employed MLR and Bayesian regularized ANNs to build semiempirical structure-property models for predicting the surface tension and liquid nanostructure of solvents containing a protic ionic liquid (PIL) with water and excess acid or base present. The results showed that all the models were successful in prediction. For IL–H2O mixture systems, Chen et al. (2022) [28] applied an ANN algorithm to build a group contribution (GC) model for predicting the viscosity of these binary mixtures at different compositions and temperatures, as demonstrated in Figure 1.2. The results show that the ANN–GC model with four or five neurons in the hidden layer is capable of providing reliable predictions on the viscosities of IL–H2O mixtures. In addition, comparisons showed that the nonlinear ANN–GC model has much better prediction performance on the viscosity of IL–H2O mixtures than that of the linear mixed model. The combination of the ANN algorithm and GC method was introduced by Fu et al. (2023) [30] to model the surface tension of IL–H2O mixtures. The results show that the model with 4, 5, and 7 neurons in the hidden layer can provide reliable predictions for the surface tension of the IL–H2O hybrid system. They also extended the ANN–GC model to the surface tension prediction of pure ILs, and the results showed that the model with 4 and 5 neurons in the hidden layer can provide reliable predictions for the surface tension of pure ILs. For IL–organic solvent mixture systems, Liu et al. (2023) [19] reported using three ML algorithms (i.e. ANN, XGBoost, and LightGBM) to build predictive models for the density and heat capacity of these binary mixtures. The results demonstrate that all three algorithms can provide accurate predictions, with the ANN model exhibiting the best performance. For using the same ML algorithms, Lei et al. (2023) [31] studied the surface tension and viscosity of IL–organic solvent mixtures. The modeling results indicate that all three algorithms can reliably predict the surface tension and viscosity of these binary mixture systems. Among them, XGBoost demonstrates the best performance for surface tension predictions, while the ANN model presents the best predictions for heat capacity. Most recently, Chen et al. (2024) [29] conducted a comprehensive modeling study on the viscosity, density, heat capacity, and surface tension of IL–IL binary mixtures. In their work, three ML algorithms, ANN, XGBoost, and LightGBM, were used. The results indicated that the ANN-based model exhibits the best prediction performance for viscosity, density, and heat capacity, while the XGBoost-based model provides the highest accuracy in surface tension predictions, as shown in Figure 1.2. Unlike IL-involved systems, the modeling study on ATPS using ML algorithms is still limited. Mohsen et al. (2020) [32] reported using SVM and ANN to build models for the prediction of phase equilibria in poly(ethylene glycol) (PEG) + sodium phosphate ATPS. The average absolute error of SVM with the differential evolution (DE) algorithm for the testing dataset is 6.71%, which, for the considered process, is in an acceptable interval. Chen et al. (2022) [33] combined the ANN algorithm with the GC method to conduct a comprehensive modeling study on the phase equilibria behavior of IL-based ATPSs, as illustrated in Figure 1.3. The ANN–GC model was trained and tested by 17 449 experimental binodal data points covering 171 IL-ABS at different temperatures (278.15–343.15 K). The results indicate that this ML-based model is capable of predicting the phase equilibria behavior of IL-ATPS in a general way. For the ATPSs involving biomolecules, Pazuki et al. (2010) [35] applied an ANN algorithm to model the partition coefficients of some well-known biomolecules (e.g. α-amylase, β-amylase, and albumin) in PEG-dextran ATPSs. The network topology is optimized, and the (6-1-1) architecture is found using the optimization of an objective function with the sequential quadratic programming (SQP) method for 450 experimental data points. The results obtained from the neural network of the partition coefficients of biomolecules in polymer–polymer ATPSs were compared with those from the modified Flory–Huggins model. Comparisons showed that the ANN is 50% more accurate than the Flory–Huggins model in obtaining partition coefficients of biomolecules in polymer–polymer ATPSs. Figure 1.2 Plots of experimental and predicted values from the GC model using ANN, XGBoost, and LightGBM in the predictions of (a) viscosity, (b) density, (c) heat capacity, and (d) surface tension of IL–IL binary mixtures. Source: [29]/with permission of John Wiley & Sons. Figure 1.3 Conceptual structure of ionic liquid-based aqueous biphasic systems combining GC method. Source: [34]/with permission of American Chemical Society. In 2023, Chen et al. [36] conducted an ANN modeling on the polymer-electrolyte aqueous two-phase systems involving biomolecules. In their work, 11 998 experimental binodal data points covering 276 polymer-electrolyte ATPS at different temperatures (273.15–399.15 K) and 626 experimental partition data points involving 22 biomolecules in 42 polymer-electrolyte ATPSs at different temperatures (283.15–333.15 K) were included. An ANN–GC model (ANN–GC model1) was built to predict the binodal curve behavior of polymer-electrolyte ATPS, while another ANN–GC model (ANN–GC model2) was developed to predict the partition of biomolecules in these biphasic systems. The modeling results show that ANN-GC model1 can give reliable predictions on the binodal curve behavior of polymer-electrolyte ATPS and ANN–GC model2, to some extent, is capable of predicting the partition coefficient of biomolecules in these biphasic systems in a widespread way. In addition, the obtained results also indicate that the tie-line length of polymer-electrolyte ATPS calculated from ANN–GC model1 can be directly used in ANN–GC model2 for predicting the partition coefficient of biomolecules in these ATPSs. Solvent screening is crucial for optimizing chemical processes. Different solvents can significantly impact reaction rates, yields, and selectivity. The choice of solvent has a direct impact on the quality and purity of the final product. The environmental impact of a process is closely tied to the choice of solvents. Solvent properties influence safety considerations in a laboratory or industrial setting. Different solvents have varying costs, and solvent selection can significantly impact the overall cost of a process. Efficient solvent screening helps in identifying the most suitable solvent for sustainable and responsible manufacturing practices [37]. However, the vast number of available solvents with diverse chemical properties makes comprehensive screening challenging. Access to accurate and comprehensive solvent property data is crucial for effective screening [38]. In some cases, data for certain solvents may be limited, affecting the reliability of the screening process. Solvent performance can vary under different process conditions. Screening solvents under various temperature, pressure, and concentration conditions adds complexity to the screening process. Balancing diverse criteria (e.g. process performance, cost, and environmental impact) to identify the most suitable solvent can be complex. There are often trade-offs between different desirable solvent properties. Finding a solvent that meets all criteria without compromises can be challenging, requiring careful consideration of priorities [39]. Effective solvent screening is essential for achieving optimal process outcomes, but it requires a balance between the complexity of the screening process and the need for accurate and reliable results. However, traditional experimental solvent screening is generally time and resource intensive. Exploring a wide range of solvents through experimentation may not be feasible within practical timeframes [40]. On the other hand, the optimal design of compounds through manipulating properties at the molecular level is often the key to considerable scientific advances and improved process systems performance, as reported in Ref. [41]. Property models are particularly essential due to their role as the foundation for the development of computer-aided molecular design (CAMD) methods [42]. The application of AI techniques to optimize and discover novel solvents for various industrial processes has gained prominence [43]. This is due to AI algorithms, particularly ML models, that can analyze vast databases of chemical information, properties, and reactions. This aids in predicting and identifying solvent candidates with specific desirable properties. ML models can predict the properties of solvents, such as viscosity, boiling point, density, and environmental impact. This helps researchers select solvents that meet the requirements of a particular application. AI is often integrated with computational chemistry methods to predict molecular interactions and behaviors, aiding in the understanding of how solvents interact with other chemicals [44]. To date, some efforts have been made to integrate AI techniques into solvent design. Liu et al. (2021) [45] proposed a machine learning-based atom contribution (MLAC)-CAMD framework for solvent design. In their work, three-dimensional atomic descriptors are used to develop an MLAC method for fast and accurate predictions of molecular ρ(σ) for the COSMO-SAC model. Then, the MLAC method is integrated with the CAMD problem, which is formulated as a mixed-integer nonlinear programming (MINLP) model and solved by a decomposition-based solution algorithm. The proposed framework was successfully used in the solvent design for the improved crystallization operation. Wang et al. (2022) [46] proposed a new CAMD approach for solvent design by combining ML algorithms with deterministic optimization. Variational autoencoder (VAE), a powerful generative ML method, is used to transfer a molecular structure into a continuous latent vector with an encoder and to convert the latent vector back to the molecule with a decoder. Solvent properties of interest are estimated by an FNN using the latent vector as input. The proposed design method successfully generated superior separation performance solvent candidates for the separation of 1-butene and butadiene. Sui et al. (2022) [47] combined eight ML algorithms with CAMD technique to design high cycle performance IL solvent for absorption heat transformer. The results showed that the optimum ILs screened by this ML-based CAMD method perform better than the currently investigated ILs. Wang et al. (2021) [48] integrated the SVM algorithm into the IL solvent design for CO2 separation from flue gas. By using this ML-based design method, 1-ethyl-3-methylimidazolium tricyanomethanide ([EMIM][TCM]) was identified as the most suitable solvent for CO2 separation from flue gas, and the performance of this IL solvent was further validated via rigorous process simulation in Aspen Plus. The results showed that the process using [EMIM][TCM] has a 12.9% savings on total annualized cost compared to that of a reported IL, 1-ethyl-3-methylimidazolium bis(trifluoromethylsulfonyl)amide ([EMIM][Tf2N]). Zhang et al. (2021) [49] presented an ML-based approach combining multiplayer Monte Carlo tree search and recurrent neural network for the tailor-made design of ILs. The application of this ML-based IL solvent design method was demonstrated through cases of CO2 capture from (i) flue gas (CO2/N2) and (ii) from syngas (CO2/H2). The results showed that the IL solvent identified through this ML-based design method presents great efficiency for the studied CO2 capture processes. Zhang et al. (2021) [50] proposed a computer-aided ionic liquid design (CAILD) approach for the optimal design of ILs for CO2 capture, where three different ML models were applied to predict the CO2 solubility. This ML-based IL solvent design problem was formulated as a MINLP problem with the objective of maximizing the CO2 solubility of ILs under prespecified conditions. The CO2 capture performance of the designed ILs was further confirmed using density functional theory calculations. The applicability of the proposed data-driven IL design method was then demonstrated via a case study of postcombustion carbon capture. Most recently, Liu et al. [51] proposed a novel ML-based IL solvent design method that combines a syntax-directed variational autoencoder (SDVAE), deep factorization machine (DeepFM), and gradient-based particle swarm optimization (GBPSO). The SDVAE converts the molecular structure and chemical space of the ILs, and then DeepFM predicts the solubility of each coordinate in the chemical space representing an IL. Finally, GBPSO identifies the coordinates that represent ILs with ideal properties. In their work, the main optimization objective was a high solubility difference for CO2 between its absorption and desorption conditions in commercial plant capture systems, which represents the CO2 capture ability. The best IL generated has a predicted solubility difference that is 35.3% higher than that of the best one in the data set. A synthetic novel IL, [EMIM][TOS], from the generated results, was experimentally evaluated; it has a sufficiently high solubility difference to be a capture solvent with low energy consumption. This model has proved to be a high-efficiency molecular design model that can be used for sparse small data sets. Kuroki et al. (2023) [52] proposed an electronic structure informatics approach to predict and develop ILs with high CO2 solubility based on geometric and electronic factors of the constituent cations and anions. With a ML-assisted search for the best cation/anion combination, targeted organic syntheses, and precision measurements, the IL trihexyl(tetradecyl)phosphonium perfluorooctanesulfonate ([P66614][PFOS]) was experimentally proven to have a higher CO2 solubility than trihexyl(tetradecyl)phosphonium bis(trifluoromethanesulfonyl)amide ([P6,6,6,14][TFSA]). Some more works associated with the ML-based solvent design of ILs for carbon capture can be found in Sun’s review study Sun et al. (2023). Chen et al. [29] combined ANN algorithms with the CAMD technique for the design of an IL–IL binary mixed solvent for recovering H2 from raw coke oven gas. The optimal solution was achieved in 30 seconds on an Intel(R) Xeon(R) E5-1620 3.70 GHz PC, demonstrating the high computational efficiency and integrability of the ANN algorithm in solvent design. Optimization results show that a binary mixed solvent combining 57.2 mol% butylethylammonium tetrafluoroborate ([N4,1,0,0][BF4]) and 42.8 mol% 1-methylimidazolium bis(trifluoromethanesulfonyl)amide ([C1Im][Tf2N]) meets all the design constraints and has the maximum H2 absorption capacity (= 0.28 g−1), as presented in Figure 1.4. To the best of our knowledge, this is the only work using ML algorithms in the design of IL–IL mixed solvents that have been reported. Figure 1.4 Molecular structure and some important properties of the binary mixed solvent combining 57.2 mol% [N4,1,0,0][BF4] and 42.8 mol% [C1Im][Tf2N]. Source: [29]/with permission of John Wiley & Sons. For ATPSs, Chen et al. (2021) [34] integrated the ANN algorithm and CAMD technique to tailor optimal IL-ATPSs for the recovery of hydrophilic ILs from dilute aqueous solutions. Two case studies were performed to test this ML-based ATPSs design method by formulating and solving their optimization-based MINLP problems. In both cases, the salting-out agents, i.e. (NH4)2SO3 and KH2PO4, identified in this work have better ABS-forming ability than their counterparts K2CO3 and (NH4)2SO4 reported in the literature. For the recovery of 10 wt% [C4Py][TfO] from aqueous solutions, the ATPS composed of [C4mIm][Cl]-H2O-(NH4)2SO3 gives an IL recovery efficiency of 95.0 wt% and a salting-out agent input of 2.36 kg/kg IL recovery, and for the ATPS of [C4mIm][Cl]-H2O-K2CO3, they are 81.7 and 5.25, respectively. For the aqueous solutions containing 10 wt% [C4Py][TfO], the ATPS composed of [C4Py][TfO]-H2O-KH2PO4 gives an IL recovery efficiency of 95.6 wt% and a salting-out agent input of 1.81 kg/kg IL recovery, and for the ABS composed of [C4Py][TfO]-H2O-(NH4)2SO4, they are 80.6 and 3.16, respectively. Most recently, Chen et al. (2023) [53] proposed an ML-based design method of high-performance ATPS for partitioning biomolecules, as presented in Figure 1.5. In this design method, two ML models that combine the ANN algorithm and GC method are, respectively, employed to predict the phase equilibrium composition of polymer-electrolyte ATPS and the partition of biomolecules in these aqueous systems. By integrating these two ANN–GC models into the computer-aided design technique, the optimal ATPS is identified by solving an optimization-based MINLP problem. Results of partitioning cefazolin and β-amylase were presented to demonstrate the viability of this ML-based ATPS design method. In the case of cefazolin, the partitioning performance of the tailored ATPS (PPG600 + KNaSO4 + H2O) is nearly seven times greater than that of the reported ATPS (PEG6000 + Na3C6H5O7 + H2O). Meanwhile, the ATPS of PPG600 + KNaSO4 + H2O gives a cefazolin recovery of 95.0 wt% and an agent input of 0.154 kg/kg aqueous solution, and for the ATPS of PEG6000 + Na3C6H5O7 + H2O, these values are 90.6 and 0.233, respectively. For the second case, the partition coefficient of β-amylase in ATPS (PPG400 + KNaHPO4 + H2O) (identified by ML-based design method) is about 13.5 times higher than that of the reported ATPS (PEG10000 + KH2PO4 + H2O). In addition, the ATPS of PPG600 + KNaSO4 + H2O gives a β-amylase recovery of 97.3 wt% at a cost of 0.387 kg agent input/kg aqueous solution, and for the ATPS of PEG6000 + Na3C6H5O7 + H2O, they are 66.3 and 0.252, respectively. Figure 1.5 Diagram of the ML-based design of ATPS for the partitioning of biomolecules. Source: [54]/with permission of American Chemical Society. Process design is a critical phase in the lifecycle of any manufacturing or industrial operation. It involves the detailed planning and configuration of systems, equipment, and processes to convert raw materials into finished products. Efficient process design ensures the optimal use of resources, including raw materials, energy, and labor. Well-designed processes are crucial for ensuring the quality and consistency of the final product. Property models play a crucial role in process design across various industries, and they are essential for sizing and designing process equipment. Parameters like heat capacity, vapor pressure, and phase equilibrium data are critical for designing reactors, heat exchangers, distillation columns, and other equipment. Property models also contribute to understanding thermodynamic behavior and ensuring the safety and environmental compliance of industrial processes. Accurate property models are indispensable for successful and efficient process design. However, there are several challenges associated with acquiring precise property models. Industrial processes often involve complex mixtures and reactions. Developing accurate property models for complex systems can be challenging due to the interactions between different components. In addition, many real-world systems exhibit nonideal behavior, such as deviations from ideal gas or solution behavior. Modeling such nonidealities accurately can be challenging. Furthermore, developing and solving complex models computationally can be resource intensive and time consuming. ML algorithms have significant potential in addressing these challenges associated with process design. This is due to the following factors: They can analyze vast amounts of process data to identify patterns, correlations, and insights that may not be apparent through traditional methods. ML algorithms can estimate properties such as viscosity, density, and thermodynamic parameters, providing accurate predictions for materials with limited experimental data. ML algorithms, particularly advanced neural networks, can capture complex relationships within systems, allowing for more accurate modeling of intricate processes. ML algorithms trained on existing data can provide estimations for properties of materials not yet extensively studied, facilitating the incorporation of new materials into process designs. Besides these, ML algorithms can integrate process models with economic considerations, optimizing designs based on both technical and economic criteria [54]. Recently, efforts have been made to integrate ML algorithms into the process design, especially for additive manufacturing. In addition to the studies discussed in Razvi’s review work, Jiang et al. (2022) [55] recently proposed an ML-integrated design framework to establish process–structure–property (PSP) relationships for additive manufacturing. With the help of ML, the analysis and design processes based on PSP no longer need to establish complex surrogate models that are also unable to establish the relationships of PSP in a reversed direction. The relationships between process, structure, and property can be established simply through ML in whichever direction it is desired using the available additive manufacturing data. DNNs for point data and CNNs for distributions and image data were proposed as the specific ML techniques for the proposed framework. Chen et al. (2023) [56] proposed an ML-based hybrid process design method for efficient recovery of hydrophilic IL from dilute aqueous solutions. The application of this ML-based hybrid process design method is illustrated through case studies of recovering two hydrophilic ILs, n-butylpyridinium trifluoromethanesulfonate ([C4Py][TfO]) and 1-butyl-3-methylimidazolium chloride ([C4mIm][Cl]), from their dilute aqueous solutions. For the recovery of 10 wt% [C4Py][TfO] from the aqueous solution, the proposed hybrid process could reduce the total annual cost (TAC) and energy consumption by 57% and 91%, respectively, compared with pure distillation processes. In the case of recovering 10 wt% [C4mIm][Cl] from the aqueous solution, the reduction in TAC and energy savings of the hybrid process could reach 49% and 87%, respectively, compared with the pure distillation process. Most recently, Chen et al. (2024) [29] integrated ML algorithms into process design involving IL–IL mixed solvent. In their work, the ANN algorithm was combined with the GC method to predict properties, including viscosity, density, surface tension, and heat capacity in the equipment design, as shown in Figure 1.6. The application of this ML-based process design method is demonstrated via a case study on H2 recovery from raw coke oven gas, as shown in Figure 1.7. In that case, the best solution was found within 27 seconds on an Intel(R) Xeon(R) E5-1620 3.70 GHz PC, indicating the high computational efficiency and integrability of ML algorithms in process design. Figure 1.6 Framework of the ML-assisted modeling, solvent tailoring, and process design involving IL–IL mixed solvent. Source: [29]/with permission of John Wiley & Sons. Figure 1.7 Process flowsheet for recovering H2 from raw coke oven gas using a binary IL–IL mixed solvent. Source: [29]/with permission of John Wiley & Sons. Despite the promising capabilities of AI techniques in property modeling, solvent tailoring, and process design, it also presents certain challenges as follows:
1
AI for Property Modeling, Solvent Tailoring, and Process Design
1.1 AI-Assisted Property Modeling
1.2 AI-assisted Solvent Tailoring
1.3 AI-Assisted Process Design
1.4 Conclusions
References