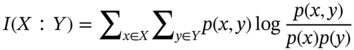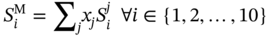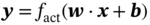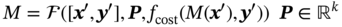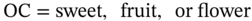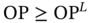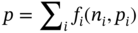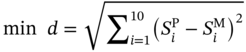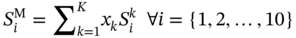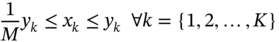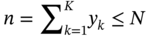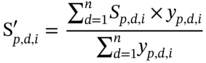Qilei Liu, Haitao Mao, Lu Wang, and Lei Zhang Dalian University of Technology, School of Chemical Engineering, Institute of Chemical Process Systems Engineering, Department of Pharmaceutical Sciences, Frontiers Science Center for Smart Materials Oriented Chemical Engineering, State Key Laboratory of Fine Chemicals, No. 2 Linggong Road, Ganjingzi District, Dalian, 116024, China Chemical-based products have always garnered significant interest in human society, as they are essential for our survival and contribute to improving our quality of life. Aromatic chemicals, in particular, are considered indispensable ingredients in various commercial products such as fragrances, shampoos, detergents, and food items. The incorporation of aromatic chemicals not only enhances the quality of these products but also sets them apart from their competitors. The global fragrance market has witnessed remarkable growth, more than doubling in size over the past 15 years, with profits soaring from US$9.6 billion in 1995 to US$22 billion in 2010 and reaching US$24 billion in 2016 [1]. The escalating demand for fragrances necessitates the development of more efficient design methods for aromatic chemicals. However, the design of aromatic chemicals is difficult due to the lack of a reliable and efficient structure–odor relationship (SOR). The development of SOR can be classified as heuristic rule-based methods and model-based methods. Among the contributions of the heuristic rule-based methods, plenty of efforts have been put into the analysis of atom location and electronic effect to draw empirical/semiempirical rules [2]. With the help of expert knowledge, the established rules show promising applications in predicting several single odors [3]. However, heuristic rules often suffer from the limitation of application range. Besides, extra conditions could undermine the heuristic rules. In addition, the development of sophisticated rules demands a long period with enormous efforts and costs. One of the examples is that the “Tommy Girl” fragrance required close to 1100 iterations before the product was released to the market [4]. Recently, researchers have started turning to model-based methods, which need efficient and general SOR models. For the odor diffusion process, the macroscopic properties are considered initially, such as partition coefficient, gas chromatographic retention times, and solubility [5]. Afterward, encouraged by the development of cheminformatics, studies of the integration of diverse categories of structural features/descriptors [6] and statistical models such as multiple linear regression are employed for the correlation of descriptors to odors [7]. One case is the development of the advanced data analysis and pattern recognition toolkit to generate seven categories of descriptors, which are further correlated to musk odor [8]. More recently, Shang et al. [9] have collected descriptors to predict gas chromatography/olfactometry of aromas, demonstrating the efficacy of model-based methods for the general prediction of various odors. However, most of the developed model-based methods are still traditional ones, such as multiple linear regression models, which often intrinsically fail to accurately model the SOR. The understanding of the odor mechanism is so difficult that simple linear equations often fail to develop general SOR models [10]. In developing general SOR models, the selection of descriptors is necessary due to increasing descriptors causing loads to the development of SOR, but it often encounters the embarrassment where these selected descriptors are based on linear models, and it is hard to explain their responsibilities to the odor at a theoretical level [11]. One of the solutions to the above challenges is to select descriptors correlated tightly with odors to formulate a more sophisticated SOR model. Lavine et al. [12] have proposed an SOR model based on the electronic van der Waals surface descriptors, which has been validated by involving biological and nonbiological molecular behavior [13]. Nevertheless, the deficiency of this type of descriptor is that it only focuses on the process of odor perception but does not consider the information on macroscopic processes such as evaporation and diffusion, which are also essential factors affecting odors [14, 15]. After the selection of descriptors, the SOR model can be established. Recently, machine learning (ML) models have often been applied to overcome the deficiencies of finding appropriate mathematical correlations of SOR in traditional statistical methods. ML is the science of autonomously learning complex relationships from data and has experienced an immensely successful resurgence during the last decade [16]. ML offers the basis of a wealth of fascinating applications, including the estimation of chemical properties [17], the prediction of organic reaction outcomes [18], and facilitating the discovery/design of catalysts, drugs, aromas, and materials [19, 20]. There are also plenty of studies for the modeling of SOR using ML models such as support vector machines [21] and random forests [22]. With the ML-based SOR model developed, aromas can be designed using computer-aided molecular design methods. The design of chemical products involves the identification of molecular structures and/or the compositions of ingredients that satisfy a set of property targets representing a set of desired product needs or attributes. In computer-aided molecular design methods, the property prediction models play an important role, as they connect the molecular structure to the desired properties. The “generate-and-test” method is widely used in computer-aided molecular design, which was proposed by Gani and Brignole [23] and then Joback [24]. It generates feasible solutions and then uses the objective function to evaluate and rank the feasible solutions. However, combinatorial explosions are likely to be encountered, especially when the design problem involves a large number of atoms. Therefore, mathematical programming-based approaches have also been developed for computer-aided molecular design. In this approach, the molecular design problem is formulated as a mixed-integer linear/nonlinear programming (MILP/MINLP) problem. Odele and Macchietto [25] have solved the optimal solvent selection problems using a mathematical programming approach. Computer-aided molecular design methods were first applied to the design of small molecules [26] and mixtures [27]. Recently, the applications have been extended to more types of products, such as amino acids [28] and ionic liquids [29]. Methods and tools are often integrated into computer-aided molecular design to expand its scope to different product design problems. For example, Austin et al. [30] have used quantum mechanics to directly predict the reaction rate in different solvents for reaction solvent design, Kupgan et al. [31] have used molecular dynamics to design polymers for CO2 capture and separations, and Guo et al. [32] have used ML methods for the discovery and optimization of additives in preparing Cu catalysts for CO2 reduction. Review articles for computer-aided molecular design methods can be found in Zhang et al. [33, 34]. Although computer-aided molecular design methods have been studied for several decades, most of the research focuses on designing small molecules such as solvents, and the design of aromas with ML methods is limited [20, 35, 36]. Therefore, the aroma design framework using ML-based SOR models is proposed. The remainder of this chapter is structured as follows: In Section 2.2, the ML methods are used to develop the SOR models for predictions of odor properties for pure and mixture aromatic chemicals. In Section 2.3, a computer-aided aroma design (CAAD) framework is proposed for designing novel pure and mixture aromatic chemicals. In Section 2.4, four case studies for the designs of aromatic chemicals are carried out using the proposed ML-based SOR models and the CAAD framework. Here, the odor pleasantness (OP) and odor characters (OCs) are selected as the required key properties for a fragrance product, which are defined as follows. The odor pleasantness is a scale from the rating of people for a certain molecule, from 0 to 100; the odor characters are classified in terms of the following 20 categories based on people’s perception [37], namely “edible,” “bakery,” “sweet,” “fruit,” “fish,” “garlic,” “spices,” “cold,” “sour,” “burnt,” “acid,” “warm,” “musky,” “sweaty,” “ammonia/urinous,” “decayed,” “wood,” “grass,” “flower,” and “chemical.” Therefore, a radar chart can be plotted for the representation of odor character for each molecule, as Figure 2.1 shows. These 20 categories of odor characters cover most of the odors for the design of fragrance products in the industry. Figure 2.1 Radar chart for the representation of odor characters. Source: Zhang et al. [20]/with permission of Elsevier. For each molecule in the radar chart, it can be found that there is one character among all the 20 that has the highest value, which is the key odor character. To simplify the problem, only the key odor character is reserved for each molecule. Therefore, we can say the odor of molecule “4-Ethoxybenzaldehyde” is sweet. Similarly, the odor of triacetin is sour, and the odor of p-cresol is chemical. In this section, the database developed by Keller et al. [38] is used. This database has 480 molecules, which are listed in Table 2.A.1 in Appendix A. The molecules have between 1 and 28 nonhydrogen atoms, including 29 amines and 45 carboxylic acids. Two molecules contain halogen atoms, 53 have sulfur atoms, 73 have nitrogen atoms, and 420 have oxygen atoms. The molecules are structurally and chemically diverse, and many of them have unfamiliar smells; some have never been used in prior psychophysical experiments. Next, the representation of molecular structures needs to be determined as the input of the ML model. Fragment (group)-based representation is commonly used in group contribution methods. It has been shown that the properties of a molecule can be determined with relatively high accuracy by summation of the contributions of the associated groups. This section uses group-based descriptors to develop ML models for predictions of the odor property. Here, 50 groups are selected, which are given in Table 2.A.2 in Appendix A. A convolutional neural network (CNN) is a class of deep, feed-forward artificial neural network (ANN), which consists of an input and an output layer, as well as multiple hidden layers, including convolutional, pooling, or fully connected layers. Python Keras [39] is used in this section for the development of the CNN-based SOR models. The input to the model is a 50 × 1 vector of groups, and the output is odor properties, including odor characters and odor pleasantness. The layer information is shown in Figure 2.2. In Keras, the embedding layers and the flatten layers are used for reshaping the data. The dropout layer is used to prevent overfitting. The dense layer is a fully connected layer, so the neurons in the layer are connected to those in the next layer. As shown in Figure 2.2, the established CNN model consists of a 50 × 64 embedding layer, a 47 × 128 convolutional layer, a 44 × 128 convolutional layer, a 22 × 128 max-pooling layer, a 22 × 128 dropout layer, a 2816 × 1 flatten layer, a 128 × 1 dense layer, another 128 × 1 dropout layer, and a 20 × 1 dense layer. Finally, the properties of odor characters and odor pleasantness are predicted using this model architecture, with different trained parameters. The 480 molecules in the database are tested using the established ML models. The training of the ML models is implemented on a desktop computer with Intel Core i7-7700 CPU and 16G memory. The training time is 696.8 seconds. The predicted results of odor characters and odor pleasantness are compared with the experimental data as shown in Figures 2.3 and 2.4. To account for the diverse psychophysical properties of odor pleasantness among individuals, it is not necessary to obtain continuous values. Instead, the odor pleasantness can be discretized into five levels based on the original odor pleasantness values. For example, level 1 can represent a range of 0–20, level 2 can represent a range of 20–40, and so on. This discretization approach enhances the representativeness and applicability of the model. Therefore, the prediction results shown in Figure 2.4 are also discretized into five levels. Figure 2.2 CNN layer information for SOR model. Source: Zhang et al. [20]/with permission of Elsevier. Figure 2.3 The predicted results of the 480 molecules in the database for odor characters. Source: Zhang et al. [20]/with permission of Elsevier. Figure 2.4 The comparison of experimental values and predicted results of odor pleasantness (scale from 0 to 100) for the 480 molecules in the database. Source: Zhang et al. [20]/with permission of Elsevier. From the results in Figures 2.3 and 2.4, it can be seen that both predictions of odor characters and pleasantness are accurate using the developed ML model, which is tested using the 480 molecules in the database. The average correctness of odor characters is 92.9%, while the average prediction error of odor pleasantness is 18.4%. In Figure 2.3, the black line shows the number of molecules for a typical odor character in the database, while the bar shows the correctness of the model prediction. In the database, characters “sweet” and “chemical” possess the largest number of molecules, while other ones possess smaller but sufficient numbers of molecules. In Figure 2.4, the two dashed lines indicate the acceptable range for the predicted properties, that is, the predicted property (indicated by dots) must be inside the region covered by the dashed lines. From Figure 2.4, 27 molecules are out of the acceptable range. Since the odor pleasantness experimental values are obtained from the rating of people, the data may not be quite accurate. Therefore, although most of the characters and pleasantness have satisfactory correctness, the prediction of odor characters for molecules outside the database has to be re-evaluated. Several aromatic chemicals outside the database, which are commonly used in our daily lives, are evaluated using the ML model. The evaluation results show roughly 75% correctness for molecules outside the database using the developed ML model. It is crucial to emphasize that each molecule is defined with only one odor character out of the 20 available. Consequently, molecules that possess multiple distinct smells are represented by a single odor character, which can impact the training of the CNN. This limitation is evident in the replacement of “floral” and “vanilla” with “sweet.” This finding demonstrates the existence of correlations between different odor characters, which are quantified using mutual information as defined in Eq. (2.1). The mutual information between each pair of odor characters is illustrated in Figure 2.5. where p(x) is the probability density of sample value x in a character vector X, p(x, y) is the joint probability density of sample values x and y, and I is the mutual value of characters X and Y. From this definition, the greater the value I, the more correlated the two characters. As shown in Figure 2.5, the mutual information value of “sweet” and “flower” is 0.78, while “sweet” and “fruity” is 0.85. This result shows that these characters are highly correlated. Therefore, the SOR model is acceptable for further aroma design problems. However, the prediction models can still be improved by introducing more molecules into the database, considering more detailed odor characters and more detailed molecular structures during the model development. The odor property data is from Keller and Vosshall [37]. They have provided 480 different molecules at four dilutions (“1/10,” “1/1000,” “1/100 000,” and “1/100 000”) to be tested by 49 healthy subjects. These subjects are required to evaluate molecules using a score from 0 to 100 for the perception of 22 specific odors, including 20 odor characters (edible, bakery, sweet, fruit, fish, garlic, spices, cold, sour, burnt, acid, warm, musky, sweaty, ammonia, decayed, wood, grass, flower, and chemical) and two odor perception characters (intensity and pleasantness). Their database of molecules includes not only familiar odor characters but also unfamiliar odor characters and even some odorless molecules like water. This odor database is applied in this section to establish the ML model. Figure 2.5 The heat map for mutual information of 20 odor characters. Source: Zhang et al. [20]/with permission of Elsevier. Since different social habits, living environments, and individual differences exist when obtaining the data in the odor database, the data needs a preprocessing step to obtain standard, rescaled, and pruned data. First, the data of 1/1000 dilution is selected as it possesses the largest amount in the database. Since people have inherent diversities, someone might be insensitive to one type of odor character or even to some typical molecules, which means the score of odors made by the subject is blank. Therefore, scored data is preprocessed to eliminate the influence of individual differences. Here, two heuristic rules are applied for preprocessing scores from subjects who are not insensitive to the odor types: With the above data selection steps, around 18 000 data are selected among 55 000 data points. Second, to avoid the disturbance from individual differences, data averaging has proceeded for the scores of the odor characters (details can be found in Appendix B). Then, the odor property data is normalized to [0, 1] using the min–max normalization method. To avoid overfitting, the preprocessed dataset is divided into training and validation sets. Here, k-folds cross-validation method is used [16], in which the whole dataset is divided into k (k = 5) subsets, and each subset is selected sequentially as the validation dataset, while the others are the training dataset. In this way, the model can be trained using the most representative dataset to avoid overfitting. The molecular surface charge density profiles (σ-profiles) of a molecule characterize its electrostatic polarity and charge distribution, which is determined by its molecular structure. Therefore, the σ-profiles can be used as descriptors for property prediction due to their ability to represent the molecular structure. In this section, the σ-profiles are used as descriptors to establish the SOR model using ML methods. Here, the σ-profiles of a molecule are calculated from the conductor-like screening model-segment activity coefficient (COSMO-SAC) model [40]. Figure 2.6 presents the process of odor perception for aroma molecules as well as the workflow for obtaining the σ-profiles descriptors. In Figure 2.6, an example molecule of Tyrosine (CAS No. 60-18-4) is shown in Figure 2.6a, and its odor is perceived when it binds to olfactory receptors in the human nose, which is shown in Figure 2.6d. The following two computation steps include geometry optimization and COSMO energy calculation. Then, the COSMO surface of the molecule is obtained, which is shown in Figure 2.6b. Afterward, the surface charge densities from the COSMO output are averaged, as shown in Figure 2.6c. These averaged charges are further projected into the two-dimensional spectrum (σ-profiles) (Figure 2.6e), which is used as descriptors for the prediction of physicochemical properties (Figure 2.6f). After the above steps, all charges of the molecular COSMO surface are converted into a two-dimensional spectrum σ-profiles with the range from −0.025 As the dilutions of the selected aroma data are 1/1000 from the database, which are dilute, the binary interaction of the ingredients can be neglected. Therefore, for aroma mixtures, a linear mixing rule (Eq. (2.2)) is applied for the σ-profiles descriptors, when the binary interaction between different ingredients of the mixture is neglected. A similar assumption has also been made from previous research [30, 43]. Figure 2.6 σ-Profiles descriptors for aroma molecules. Source: Zhang et al. [41]/with permission of Elsevier. Figure 2.7 σ-Profiles descriptors of an example aroma. Source: Zhang et al. [41]/with permission of Elsevier. Figure 2.8 Linear mixing rule for aroma mixtures is reasonable. Source: Zhang et al. [41]/with permission of Elsevier. In Eq. (2.2), i is the index of 10 area parameters of the descriptors, j is the index of compounds in the aroma mixture, Figure 2.8 shows the comparison of σ-profiles descriptors of two example aroma molecules and the mixture from the COSMO-SAC method and the linear mixing rule. From the comparison, it is seen that the error of the linear mixing rule for the calculation of σ-profiles descriptors for aroma mixtures is acceptable. After the selection of descriptors, the SOR model is established with the aroma mixture properties as the input and σ-profiles descriptors as the output. In this way, with a certain set of requirements for aroma design problems, the optimal aroma mixture can be designed directly with the ANN model. It should be noted that it is possible that the optimal aroma mixture from the established ML model is not unique. The design results can be further tested by experiments, which could greatly reduce the cost and manpower in the aroma design process. ANN is employed for the modeling of SOR. As Figure 2.9 shows, the aroma mixture properties including seven odor properties (edible, bakery, sweet, fruit, sour, flower, and odor pleasantness) and two physical properties (vapor pressure and diffusion coefficient) are taken as the input, and the 10 area parameters (S1, S2, …, S10) of σ-profiles are taken as the output. Here, multiple-model strategy is used, where each ANN model uses all the input parameters to predict one output (Si) separately. In Figure 2.9, each ANN model utilizes a multilayer perceptron structure. The architecture of each ANN model comprises multiple layers, with each layer consisting of a set of nodes referred to as “neurons.” The detailed architecture of each ANN model is depicted in Figure 2.10. A single neuron contains two neuron layers: one consists of the input neurons x and the other consists of the output neurons y. The connections between the inputs and outputs are called weights w, the threshold values of outputs are called biases b, and the activate function is fact. The inputs and outputs of a single neuron can be represented as Eq. (2.3). The established ANN model is then trained to obtain an optimized model using the training and validation dataset. The training process aims to minimize the cost function fcost of model M by employing the learning algorithm F, which can be represented as Eq. (2.4). Figure 2.9 Multiple-model strategy for SOR of aroma mixture design. Source: Zhang et al. [41]/with permission of Elsevier. Figure 2.10 Model architecture of one ANN model. Source: Zhang et al. [41]/with permission of Elsevier. (a) A single neutron and (b) an ANN model representation. where P is the hyperparameters for the ML model, x′ is the preprocessed input data (odor and physical properties), and y′ is the preprocessed output data (σ-profiles descriptors). The performance of the ANN model is determined by the number of hyperparameters such as neuron layers [16]. When a new layer is added to the ANN model, the previously determined parameters have to be tuned again. The hyperparameters of the ANN model P are tuned by using the grid search method. The ANN model is established and trained using Python Keras package [39] using a desktop with i7-7500 CPU, 8G memory. After determining the hyperparameters, each submodel is trained five times, and the average values are compared and analyzed, which are listed in Table 2.C.1 in Appendix C. Figure 2.11 shows the performances of 10 models in the testing dataset. The average R2 for the full model is 0.88, which is acceptable considering the distribution of samples in the testing dataset may be inconsistent with the distribution of samples in the training dataset. Hence, the established ANN model can be used for further aroma mixture design. Figure 2.11 Performance of the established ANN model. Source: Zhang et al. [41]/with permission of Elsevier. Aromatic chemicals need to be designed/screened to meet all their sensorial and technical requirements. CAAD uses mathematical modeling to determine the molecular structure that matches the given set of target properties with the given set of groups used in the property models. Here, a CAAD framework is established for the design of novel pure aromatic chemicals. In this CAAD framework, the odor of the molecules is predicted using the developed ML models, while group contribution-based models are included to predict the rest of the needed physical properties, such as vapor pressure, solubility parameter, and viscosity. The CAAD problem is formulated as an MINLP model for the design of aromatic chemicals. The decomposition-based solution approach [44] is used to obtain the optimal result. Figure 2.12 shows the steps for the development and solution of the MINLP model. The design steps of the aromatic chemicals are discussed in this section. The first step is to identify the product attributes. The aromatic chemicals are the active ingredients or the additives of the fragrances. After the application of the fragrance, the aromatic chemicals begin to evaporate into the headspace at different rates depending on their volatility, composition, and molecular interactions. Subsequently, the gas odorant molecules will diffuse through the surrounding air over time and distance, and finally, at a given time and distance, some of the aromatic chemicals will eventually reach the nose of the customer who perceives the odorants with a certain intensity and character [14, 15]. From this process, it can be seen that the odor properties, including odor character and odor pleasantness, and the physicochemical properties including diffusion, evaporation, and so on, are the required product attributes. Figure 2.12 Steps for the development and solution of the MINLP model for the design of aromatic chemicals. Source: Zhang et al. [20]/with permission of Elsevier. In the second step, the product attributes are converted to properties and represented as constraints in the MINLP model. The product attributes are converted to physicochemical properties such as diffusion coefficient (diffusion), vapor pressure (evaporation), boiling and melting points (product form), solubility parameter (solubility), viscosity and density (rheology), and LC50 (environment and health), and their constraints. These converted properties need to be estimated using appropriate predictive property models from the model library. The established ML model is used for odor characters and pleasantness, while for other properties (e.g., diffusion coefficient, vapor pressure, boiling point), group contribution method, theories for thermodynamics, and transport phenomena are used. The aroma design problem can be formulated as a MILP/MINLP optimization problem. The optimization model includes an objective function, molecular structure constraints, and property constraints. The objective function can be used to maximize/minimize one of the desired properties, such as odor pleasantness, as Eq. (2.5) shows. OP is predicted using the machine learning model, in which the groups are the input variables. The structure constraints define the molecular structure from the combination of a set of feasible groups. Therefore, the groups are selected first. For aromatic chemicals, groups containing oxygen, sulfur, and aromatic rings are commonly included, such as alcohols, ketones, esters, and ethers. The set of groups is defined as i or i′ in the optimization model. The following equations define the property constraints. The odor character (OC) should be sweet, fruit, or flower from the 20 characters, as Eq. (2.6) shows. The constraint for odor pleasantness (OP) is defined in Eq. (2.7). The aromatic chemicals should have a pleasant odor; therefore, OP should be greater than a certain value (OPL). The detailed structure constraints and other property constraints can be found in Zhang et al. [20]. From the solution of the established MINLP model, the optimal aromatic chemicals are found. However, the model contains properties that have to be obtained from ML models, and at the same time, the property model equations for diffusion coefficient and vapor pressure are nonlinear equations. The existing MINLP solvers are not able to solve the design problems. Therefore, the decomposition-based algorithm [44] is used to solve the MINLP model. In the decomposition-based algorithm, the MILP/MINLP model is decomposed into an ordered set of subproblems. Each subproblem requires only the solution of a subset of the constraints from the original set. The final subproblem contains the objective function and the remaining constraints. In this way, the solution of the decomposed set of subproblems is equivalent to that of the original MILP/MINLP problem. Here, the structure constraints, and the property constraints, except the odor character, odor pleasantness, diffusion coefficient, and vapor pressure model, are considered in the first subproblem to generate feasible candidates. Then, each molecule in the set of candidates is tested using the equations of diffusion coefficient, vapor pressure, and the ML models of odor character and pleasantness. In this way, the size of the feasible candidates set becomes smaller, and finally, the optimal one (or several candidates) can be selected based on the evaluation of the objective function. Here, the model formulation and solution of the CADD problem has been implemented into a self-developed software “OptCAMD” [45]. Figure 2.13 shows the software architecture of OptCAMD. The interface collects all the input data from the user, including structure information such as group selection, group numbers, and all the property constraints. Then the interface transforms and reformulates all the input data to an Excel file, which is needed in the GAMS code template. The GAMS code template calls the solver to solve the optimization problem and generates a list of feasible groups based on the ranking of the objective value. A molecular database (OptCAMD database) is integrated into the software, which contains around 10 000 commonly existing molecules. The generated groups are compared with the molecules in the database, and feasible molecules are returned. These molecules can be imported to ICAS ProPred [46] and PubChem database [47] for verification. Finally, the generated molecules are displayed in the interface. The user can also modify the GAMS code template to customize the optimization problem. In this step, the obtained aromatic chemicals from the optimization model are verified through database search and/or experiments. If the designed molecules are known and exist in the database, then their measured property values can be verified from the database, or use of rigorous property prediction models, such as ICAS [46] and VPPD-LAB [48]. If the designed molecules cannot be found in any database, that means they are new molecules or their properties have not been measured yet, but they could be used in aroma products. Therefore, experiments are recommended to verify their properties and usability. In this section, a CAAD framework is proposed for the design of a novel mixture of aromatic chemicals with the ML-based SOR models developed in Section 2.2. Figure 2.14 shows the proposed CAAD framework for mixture aroma design. The steps of the CAAD framework are discussed in detail in the following text. Figure 2.13 Software architecture of OptCAMD. Source: Zhang et al. [20]/with permission of Elsevier. Figure 2.14 Computer-aided aroma design for novel mixture aromatic chemicals. Source: Zhang et al. [41]/with permission of Elsevier. The first step of the CAAD framework is identifying the product attributes. Here, a set of requirements are collected and determined according to the product application background. The requirements could be categorized into chemical engineering-related, sensorial-related, and regulatory attributes. For example, in candle fragrance products, the aroma is designed with the technical requirements of phase stability, evaporating and diffusing properties, duration, etc. The sensorial-related attributes include requirements of odor characters and pleasantness of the aroma, which determine the product performance. Furthermore, the regulatory requirements, including toxicity, safety, and so on, should be considered. These product attributes are usually collected by the market study of potential consumers. After identifying the product attributes, they are converted into properties such as flash point, boiling and melting points, vapor pressure, diffusion coefficient, vapor pressure, LC50, etc., which provide the translation from the marketing specialists to the R&D engineers. These product attributes are converted to properties, which can be predicted by theoretical property models, ML models, and/or obtained by experiments, with corresponding constraints. The boundaries of these constraints are obtained by satisfying the expectation of the product attributes, which are normally derived from consumer surveys or reference data from available products. The property models are established in this step to correlate the product structure/mixture ingredient composition with the properties. According to different properties and their available models, different property prediction models are selected. For example, group contribution methods [26] are often applied to some common properties such as normal boiling point, critical properties, and so on, as Eq. (2.8) shows. In Eq. (2.8), p is the properties, ni is the number of group i, and pi is the contribution of group i in property p, fi is a certain function in group contribution methods, which is different in different properties. For property models that are not available, but for which a large number of data exists, ML models can be established. Examples include the SOR model established in Section 2.2. If only limited experimental data exists, quantitative structure–property relationship models can be established by regressing the experimental data. For aroma mixture design problems, the σ-profiles descriptors (S1 ∼ S10) are obtained from the input properties using the established ANN model. After the σ-profiles descriptors (S1 ∼ S10) obtained, the ingredients of the mixture and their compositions need to be screened to find the corresponding aroma mixture that fits the σ-profiles descriptors. The ingredients are searched from the odor database [37], which has 480 molecules. The σ-profiles descriptors of the mixture are calculated using Eq. (2.2) if the ingredients and their compositions are known. In this step, Euclidean distance is used to evaluate the difference between the σ-profiles descriptors of the designed mixtures and the predicted values, which is shown in Eq. (2.9). where d is the Euclidean distance between the σ-profiles descriptors of the designed mixtures and the predicted values, which should be minimized. where xk is the volume fraction of ingredient k and K is the number of molecules in the odor database, which equals 480 in this section. The number of selected ingredients n should follow the constraints of Eqs. (2.11) and (2.12). In Eq. (2.11), M is a big number and yk is a binary variable that indicates whether compound k is selected as the ingredient of the aroma mixture. If yk = 1, The property constraints such as solubility, boiling point, log(Ko/w), LC50, flash point, and so on are also considered for the screening of aroma mixtures to guarantee the product performance, as shown in Eq. (2.13). where pL and pU are the lower and upper bounds of property p, respectively. pk is the property value of ingredient k for property p and f is the function of the mixing rule. An MINLP model is formulated for the ingredient screening of the optimal aroma mixture, in which Eq. (2.9) is the objective function, Eqs. (2.10)–(2.13) are the constraints. The decomposition-based solution approach [44] is applied to the solution of the optimization model. From the solution of the MINLP model, the ingredients and the volume fractions of its ingredients are obtained. It should be noted that the above mathematical model will always have feasible solutions, but the generated solution may be too far away from the required σ-profiles. In this situation, the odor database can be expanded to include a larger number of molecules. Finally, the designed aroma mixtures are verified in the verification step. The verification of some properties can be performed using a database if the property data are available in the database. For odor properties, experimental verification is needed. An electronic nose is used to verify the odor properties. Besides, consumer testing studies can be applied to obtain more accurate verification of the design results after the product prototypes are made. Based on the CAAD framework introduced above, four case studies for designing aromatic chemicals are presented in this section. Aromatic ingredients are commonly used in products such as shampoos, shower gels, shaving creams, and body lotions. Even some products labeled “unscented” may contain aromatic ingredients. This is because the manufacturer may add just enough aromas to mask the unpleasant smell of other ingredients, without giving the product a noticeable scent. On the other hand, the aromatic ingredient should meet other technical requirements. The objective of this case study is to find suitable aromatic chemicals as additives for shampoo using the developed MINLP model. According to the above analysis, the aromatic chemicals for shampoo additives should have the following properties. Table 2.1 shows the properties and constraints of the aromatic chemicals in this case study. Groups CH3, CH2, CH, C, CH2=CH, CH=CH, CH2=C, CH=C, OH, CH3CO, CH2CO, CH3COO, CH2COO, CH3O, and CH2O are selected. The MINLP model for the case study is established based on their upper and lower bounds in Table 2.1. The decomposition-based algorithm is used for the solution of the MINLP model, as Figure 2.15 shows. Table 2.1 Properties and constraints for the design of aromatic chemicals. Figure 2.15 The decomposition-based algorithm for the aroma design case study. Source: Zhang et al. [20]/with permission of Elsevier. First, feasible candidates are generated by matching constraints Tb, Tm, Sp, η, ρ and −log(LC50) as the model equations for these properties are linear. 40 feasible molecules are generated in this subproblem using the OptCAMD software. Then, constraints D and Psat are added to evaluate each generated candidate to check if they satisfy these additional constraints. 26 molecules are selected in this subproblem. Then, the 26 molecules are tested using the ML model for odor character prediction to test if these molecules are “sweet,” “fruit,” or “flower” (as defined in Table 2.1), and 8 molecules are found to match these constraints. The odor pleasantness model is then used for the screening of these eight molecules, which finds six molecules matching this constraint. The final solution is the molecule that has the highest odor pleasantness within these six molecules. The six generated molecules satisfying all property constraints are listed in Table 2.2, together with their properties. From the optimization result, molecule C9H18O2 has the highest odor pleasantness. Therefore, it is selected as the best potential aroma in this case study. Database search has been made for all the six feasible molecules. The optimal molecule, however, is not found in any database as aromas and, therefore, it needs to be evaluated through experiments to verify if the odor properties are the same as predicted. The molecules C8H16O (CAS No. 111-13-7) and C8H16O2 (CAS No. 106-73-0) are found in the database as commonly used aromas for various purposes, which confirms the effectiveness of the CAAD framework. An insect repellent spray usually constitutes an active ingredient (repel mosquitoes), a binary solvent mixture (deliver the active ingredient on the skin and vaporize after application), and additives (e.g., perfumes, moisturizing agents, etc.) [49]. It is designed with high effectiveness against mosquitoes, a water-based spray, pleasant scent, long durability, low toxicity, high stability, spray-ability, low price, and long shelf life. Picaridin is selected as the active ingredient for repelling mosquitoes by using a knowledge-based design method. However, the solubility of Picaridin in water is rather low. Therefore, a water–organic solvent mixture is an alternative to be used in order to dissolve Picaridin. A mixture of water and 2-propanol is designed as the solvent by Conte et al. [49] by using a computer-aided molecular design method for mixtures. Aromas should also be added to obtain a pleasant scent. The ingredient composition of the insect repellent spray is given in Table 2.3. Table 2.4 shows the properties and constraints of the aromatic chemicals in this case study. The objective of this case study is to design the aroma. Table 2.2 The generated feasible candidates. Table 2.3 Ingredient composition of the insect repellent spray. Table 2.4 Properties and constraints for the design of aromatic chemicals. Groups CH3, CH2, CH, C, CH2=CH, CH=CH, CH2=C, CH=C, OH, CH3COO, CH2COO, and CHCOO are selected. According to the previous analysis, the aroma should have the following physical properties, as listed in Table 2.4. The MINLP model for the case study is established based on their upper and lower bounds in Table 2.4. The decomposition-based algorithm is used for the solution of the MINLP model. Constraints Mw, Fp, Tb, Tm, Sp, η, Vm, and −log(LC50) are first used to generate feasible candidates by using the OptCAMD software. A total of 132 feasible molecules are generated in this subproblem. Then, constraints D and Psat are added to evaluate each generated candidate, and 44 molecules are found to satisfy these two additional constraints. Then, the 44 molecules are tested using the odor character prediction model to test if these molecules are “sweet,” “fruit,” or “flower,” and 16 molecules are found to satisfy this requirement. The odor pleasantness model is then used for the screening of the remaining 16 molecules, which leads to 15 molecules. The final solution is the molecule that has the highest odor pleasantness among the 15 molecules. Details of the generated 15 molecules, together with their properties, are given in Table 2.D.1 in Appendix D. From the results in this table, the ninth molecule (Linalool, CAS No. 78-70-6) is found in the OptCAMD database, and the predicted physical properties as well as odor characters agree well with the available measured data. Therefore, Linalool is selected as the aroma in this product. The selection agrees with the heuristic selection reported by Conte et al. [49]. The experimental verification is conducted, and the properties and phase stability are tested by Conte et al. [49]. With the increasing applications of aromas in chemical products such as fragrances, shampoos, cosmetics, etc., the design of aromas with consumer requirements becomes essential. The aroma cis-3-hexenyl propionate (CAS No. 33467-74-2) is a typical aroma applied widely, which smells fruity. In this case study, aroma mixtures with similar odor and physical properties with cis-3-hexenyl propionate are designed at a cheaper price. These product attributes are converted into properties that are listed in Table 2.5. Here, the property values are obtained from cis-3-hexenyl propionate. Based on the property values of edible, bakery, sweet, fruity, flower, sour, pleasantness, vapor pressure, and diffusion coefficient, the established ANN-based SOR model is used to obtain the σ-profiles descriptors of the designed mixture. The result is shown in Figure 2.16. Table 2.5 Properties of cis-3-hexenyl propionate. a) Data from the odor database [37]. b) Data from the Good Scents Company Information System (http://thegoodscentscompany.com). c) Data from group contribution methods in ICAS (https://www.pseforspeed.com/icas). Figure 2.16 ANN-predicted results using the σ-profiles descriptors. Source: Zhang et al. [41]/with permission of Elsevier. Then, the established MINLP model for ingredient screening is applied to search for feasible aroma mixture candidates, which considers the property constraints listed in Table 2.6. The compounds in the database for ingredient screening are listed in Table 2 E.1 in Appendix E. The number of ingredients is set to two. Finally, the rank of the feasible aroma mixtures based on the Euclidean distance is shown in Figure 2.17. The red line in Figure 2.17 is the tolerance of the Euclidean distance, which is set to 0.0035 in this case study. Among the aroma mixture candidates, the ingredients of the second best one (CAS No. 97-61-0 and 7493-69-8) are all commonly used aromas, which are much cheaper than cis-3-hexenyl propionate. Therefore, the second best one is used for further verification. Table 2.7 lists the properties of the ingredients of the designed aroma mixture. From the results, it is seen that all property requirements are met, and the price of the designed aroma mixture is much cheaper than cis-3-hexenyl propionate. Finally, the designed aroma mixture is verified by the electronic nose system (PEN3, Airsense Analytics GmbH, Germany) (E-nose). The samples of cis-3-hexenyl propionate and the designed aroma mixture are placed in two airtight 25 ml vials at 25 ± 1 °C for approximately 30 minutes, and odors are measured for 5 minutes at 1 second intervals. All samples are run with three repetitions and average values are obtained for the two samples. The E-nose results of the two samples are shown in Figure 2.18. Table 2.6 Property constraints of the case study. Figure 2.17 The rank of the feasible aroma mixtures based on the Euclidean distance. Source: Zhang et al. [41]/with permission of Elsevier. Table 2.7 Ingredient properties of the designed aroma mixture. a) The Good Scents Company Information System. b) ICAS. c) Sigma–Aldrich (https://www.sigmaaldrich.com). Figure 2.18 E-nose experimental results of cis-3-hexenyl propionate and the designed aroma mixture. Source: Zhang et al. [41]/with permission of Elsevier. The results indicate the odor of the two samples is almost the same. Then, human tests are also conducted. From the tests, cis-3-hexenyl propionate smells like grass, and after evaporation for a certain time, the odor switches to fruity slightly. The designed aroma mixture smells like grass, which is quite similar to cis-3-hexenyl propionate. Therefore, from the experimental results, the effectiveness of the proposed CAAD framework and the ML model is verified. The tuning of the odor of aromas is also an interesting topic in aroma design. On the one hand, some types of pleasant odors can be enhanced to improve the quality of the product. On the other hand, some unpleasant odors can be covered through the tuning of odor. In this case study, the proposed CAAD framework and the ML model are applied for the odor tuning of aromas. Here, cis-4-heptenal (CAS No. 6728-31-0) is taken as an example, which is often used as a creamy fragrance. This aroma is tuned by the design of aroma mixtures with improved edible, sweet, and fruity odors to enhance its quality. Table 2.8 summarizes the properties and the tuning targets of cis-4-heptenal. Table 2.8 Properties of cis-4-heptenal and the target values of the odor tuning. a) The Good Scents Company Information System. b) ICAS. Table 2.9 Ingredient properties of the designed aroma mixture. a) The Good Scents Company Information System. b) ICAS. c) Sigma–Aldrich. With the same design steps, the aroma mixture design results are listed in Table 2.9 and Figure 2.19. From the results of Table 2.8, it is seen that all properties meet the requirements, and from Figure 2.19, the odors of edible, sweet, and fruit of the designed mixture are enhanced, which means the requirements of the design target. The price of the designed mixture (940.69 × 0.9 + 41 179.3 × 0.1 = 4964.55 CNY kg−1) is also much cheaper than cis-4-heptenal (38 958.64 CNY kg−1). Figure 2.19 Radar plot of the odor properties of the designed aroma mixture and cis-4-heptenal. Source: Zhang et al. [41]/with permission of Elsevier. Modern society has growing demands for aromas. Since odor properties are essential for aroma design, the SOR has been proposed for better understanding the process of odor perception and further discovering potential aromas. The model-based aroma design method is utilized in this chapter because of its efficiency in SOR development and general application. The descriptors of groups and σ-profiles are used to develop SOR models for predictions of odor properties of pure and mixture aromatic chemicals using ML algorithms. The accuracy of the SOR model for pure aromatic chemicals is 92.9%, while the R2 of the SOR model for mixture aromatic chemicals is 0.88, showing the high accuracy of the established SOR models in predicting odor properties. Subsequently, a CAAD framework for designing novel pure and mixture aromatic chemicals is proposed. Four case studies are presented and verified by experiments and literature data, the results of which demonstrate the feasibility and effectiveness of the CAAD framework as well as the established ML models. Table 2.A.1 CAS registry number of the fragrance molecules in the database. The scores of all odor characters for one molecule are averaged as shown in Eq. (2.B.1): where Sp, d, i is the score of property p for the molecule i from a subject d, yp, d, i is a binary value, which is assigned to 0 or 1 for whether it is eliminated. Table 2.A.2 Selected groups for the representation of aromatic molecules. Table 2.C.1 ANN model parameters and result analysis. Table 2.D.1 The generated feasible candidates for case study 2. Table 2.E.1 Aroma compounds for ingredient screening. The authors are grateful for the financial support of NSFC (22078041, 22278053), and “the Fundamental Research Funds for the Central Universities” (DUT22YG218).
2
Hunting for Better Aromatic Chemicals with AI Techniques
2.1 Introduction
2.2 Machine Learning-Based Odor Prediction Models
2.2.1 Odor Predictions for Pure Aromatic Chemicals Using Group-Based Machine Learning Method
2.2.1.1 Database Preparation
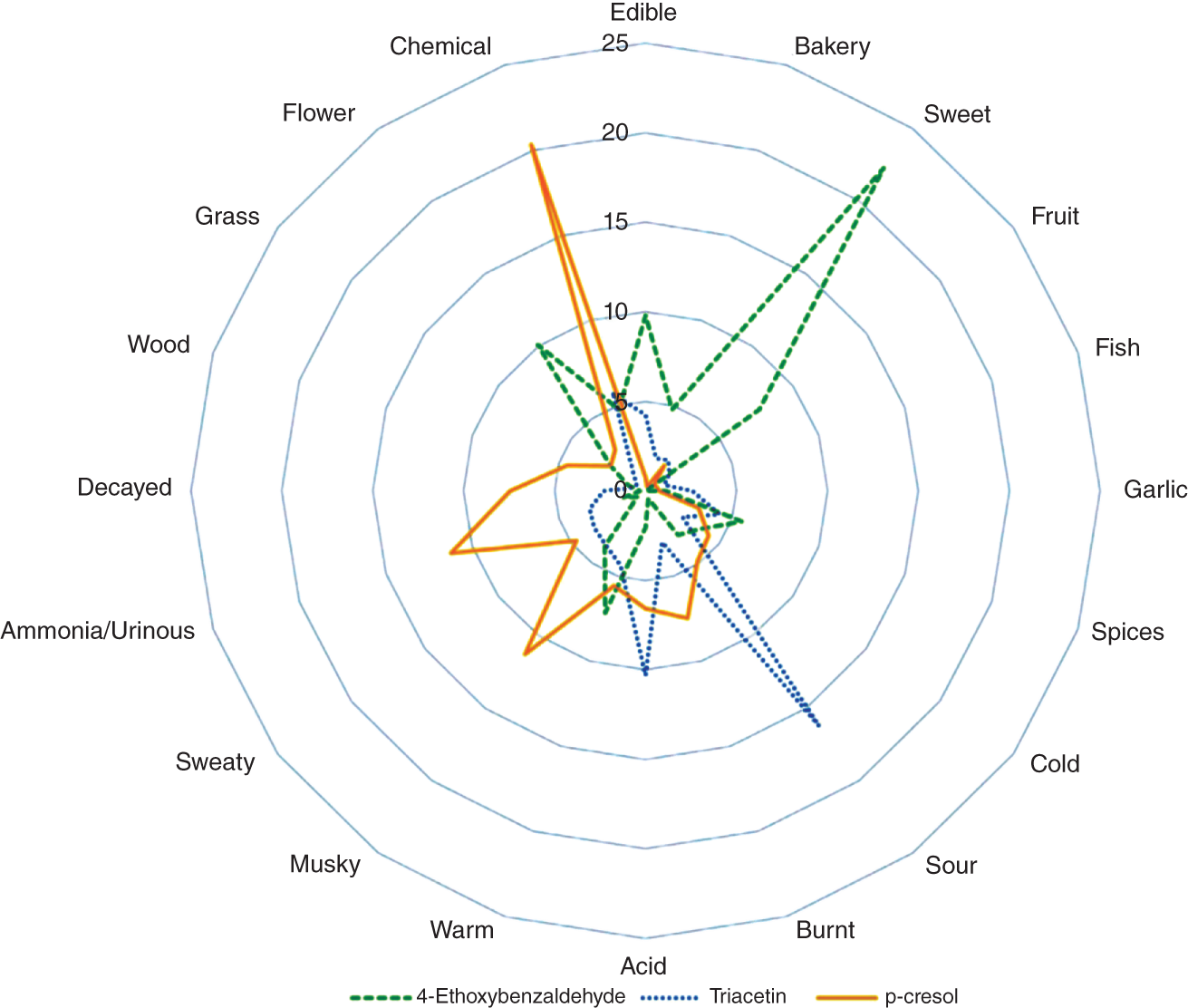
2.2.1.2 Molecular Representation
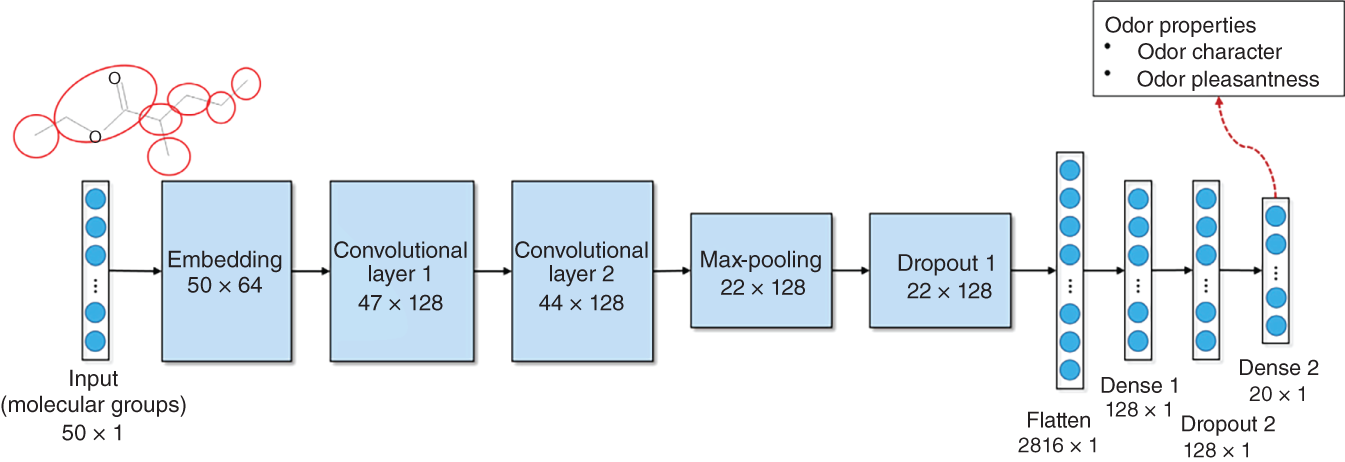
2.2.1.3 Model Architecture
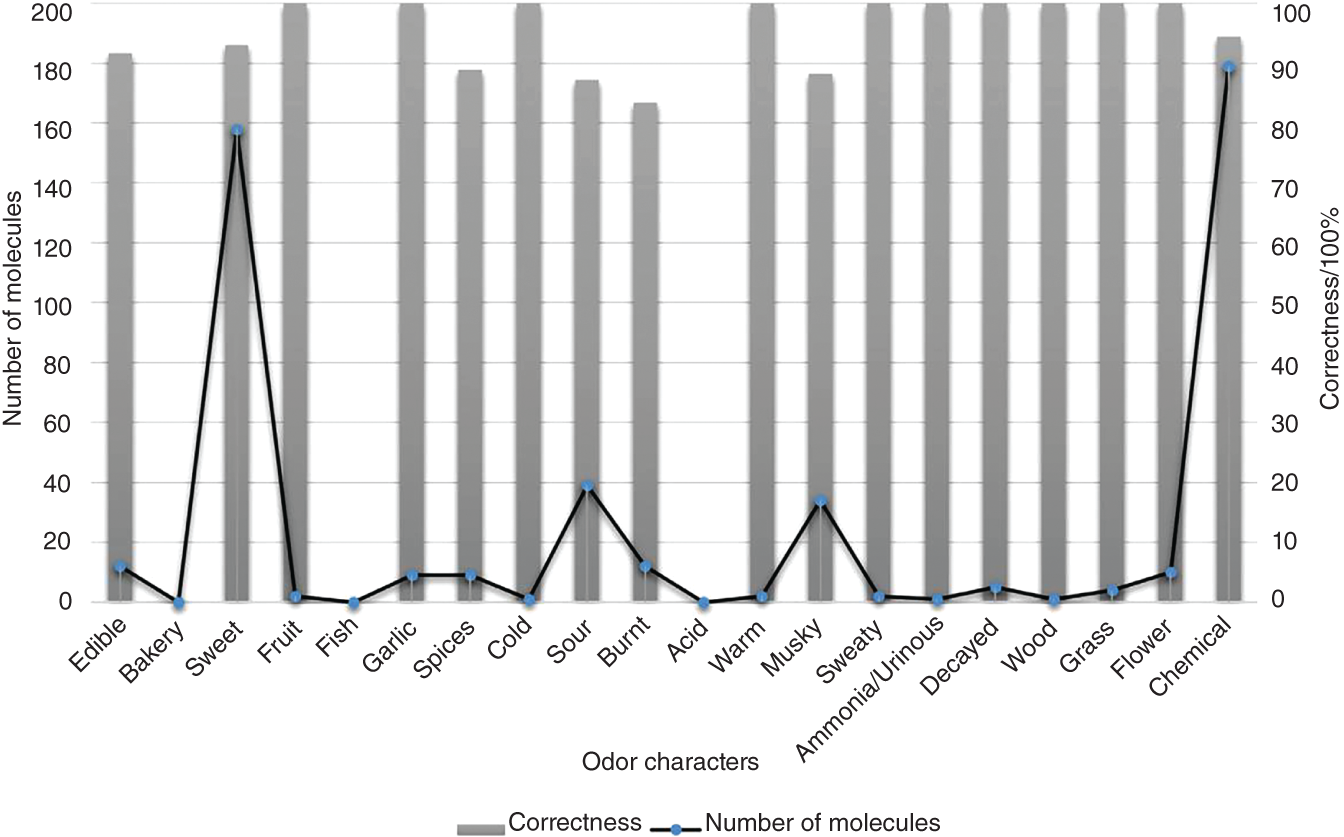
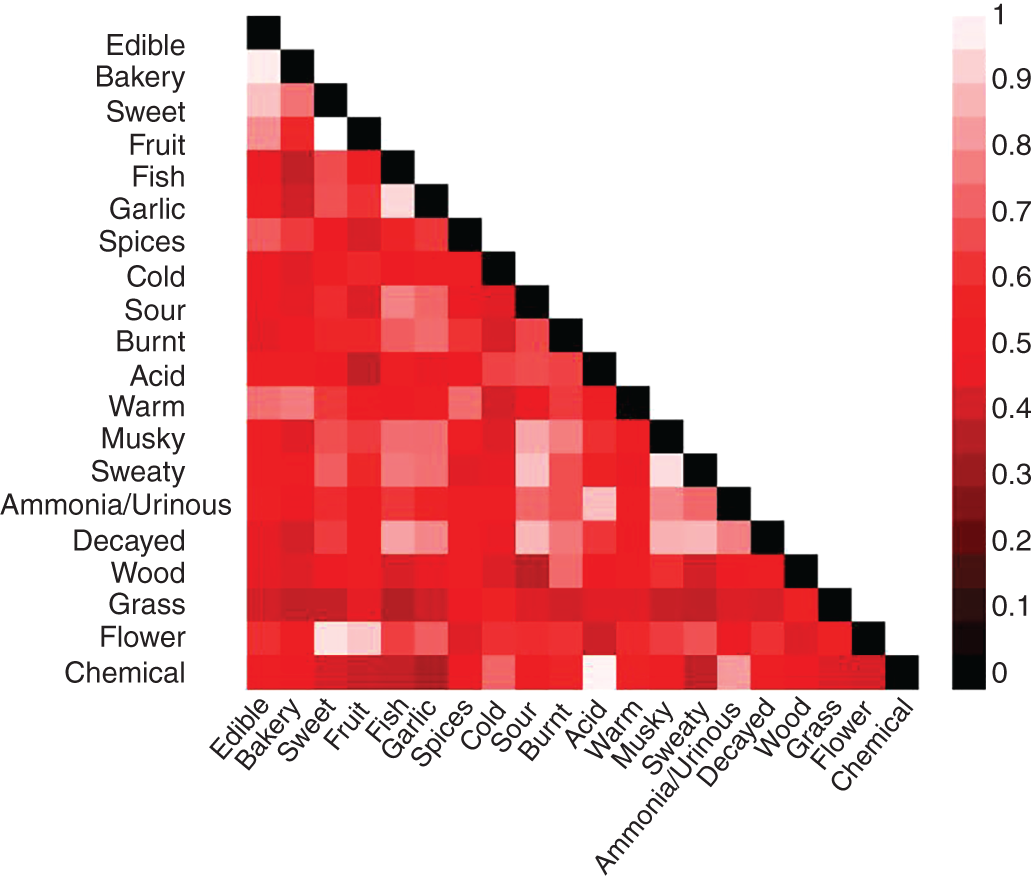
2.2.1.4 Results and Discussions

2.2.2 Odor Prediction for Mixture Aromatic Chemicals Using σ-Profiles-Based Machine Learning Method
2.2.2.1 Database Preparation
2.2.2.2 Molecular Representation
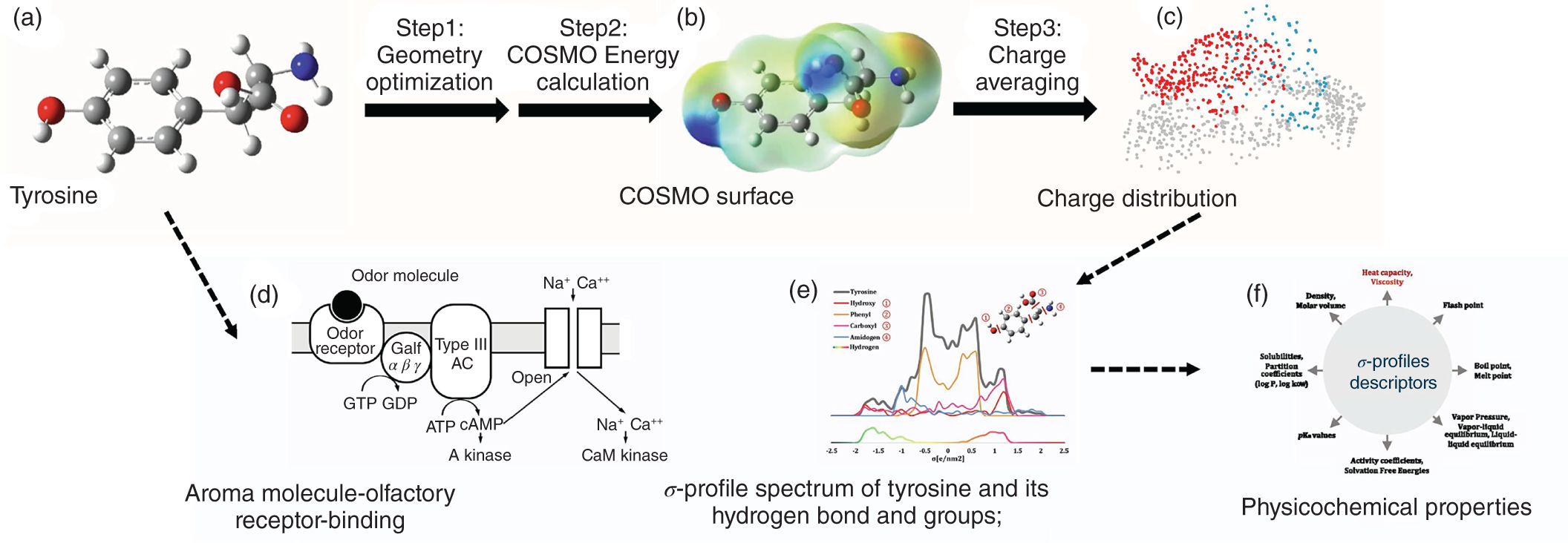
 (−2.5 e nm−2) to 0.025
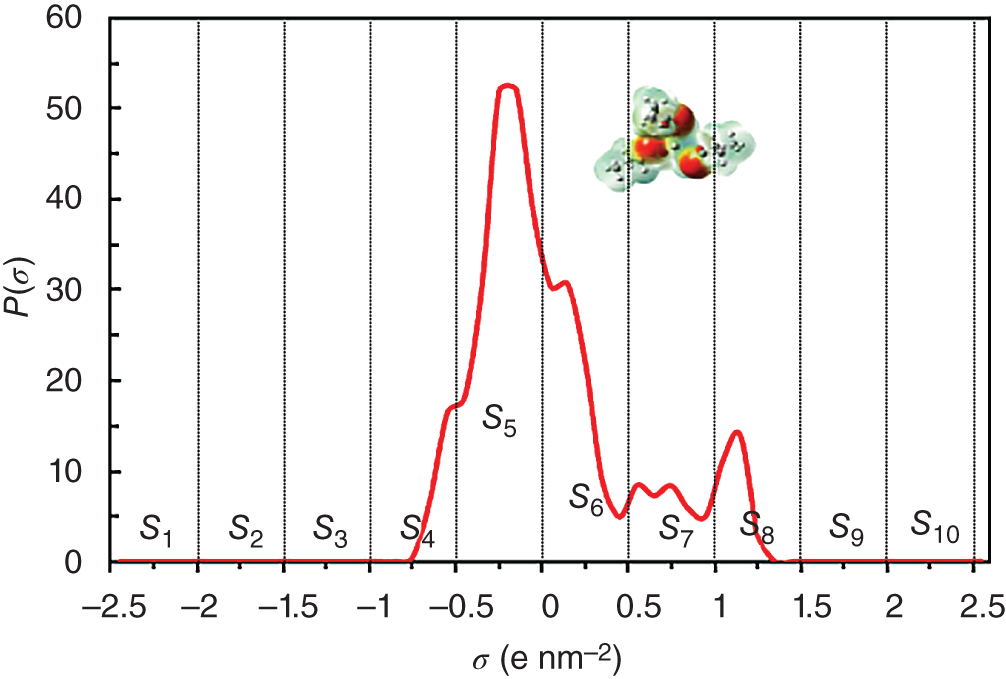
(−2.5 e nm−2) to 0.025  (2.5 e nm−2) with 51 data points in total. As shown in Figure 2.7, the σ-profiles of an aroma molecule are divided into 10 segments in the entire σ region. Thus, 10 areas (S1–S10) are obtained for each molecule by integrating those segments [42]. Afterward, the 10 area parameters are used as descriptors to quantify their effects on the odor properties. It is noted that min–max normalization is then employed to scale the σ-profiles descriptors (S1–S10) to [0, 1] to ensure all features (descriptors) have the same scale and for the purpose of obtaining the global optimum [16].
(2.5 e nm−2) with 51 data points in total. As shown in Figure 2.7, the σ-profiles of an aroma molecule are divided into 10 segments in the entire σ region. Thus, 10 areas (S1–S10) are obtained for each molecule by integrating those segments [42]. Afterward, the 10 area parameters are used as descriptors to quantify their effects on the odor properties. It is noted that min–max normalization is then employed to scale the σ-profiles descriptors (S1–S10) to [0, 1] to ensure all features (descriptors) have the same scale and for the purpose of obtaining the global optimum [16].
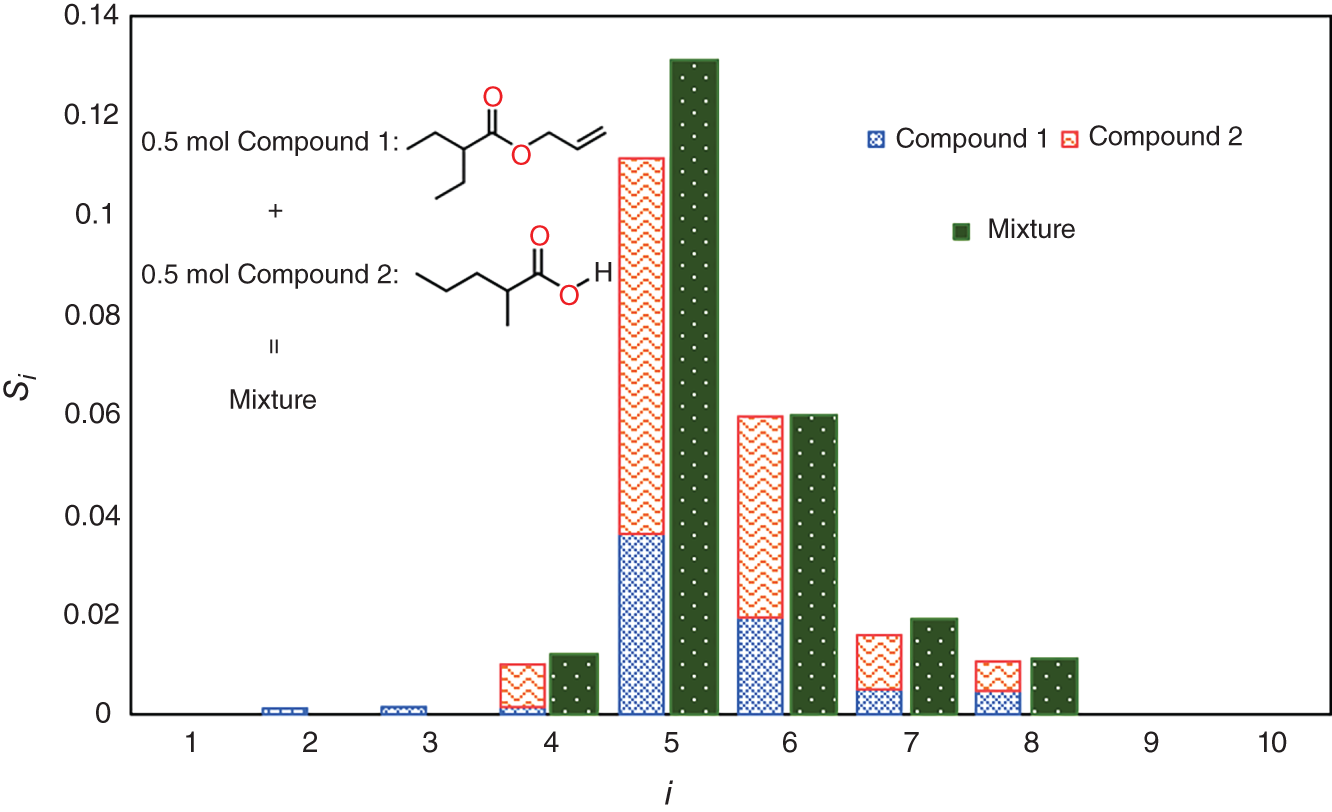
 is the ith area parameter of the mixture, xj is the volume fraction of compound j, and
is the ith area parameter of the mixture, xj is the volume fraction of compound j, and  is the ith area parameter of compound j.
is the ith area parameter of compound j.
2.2.2.3 Model Architecture
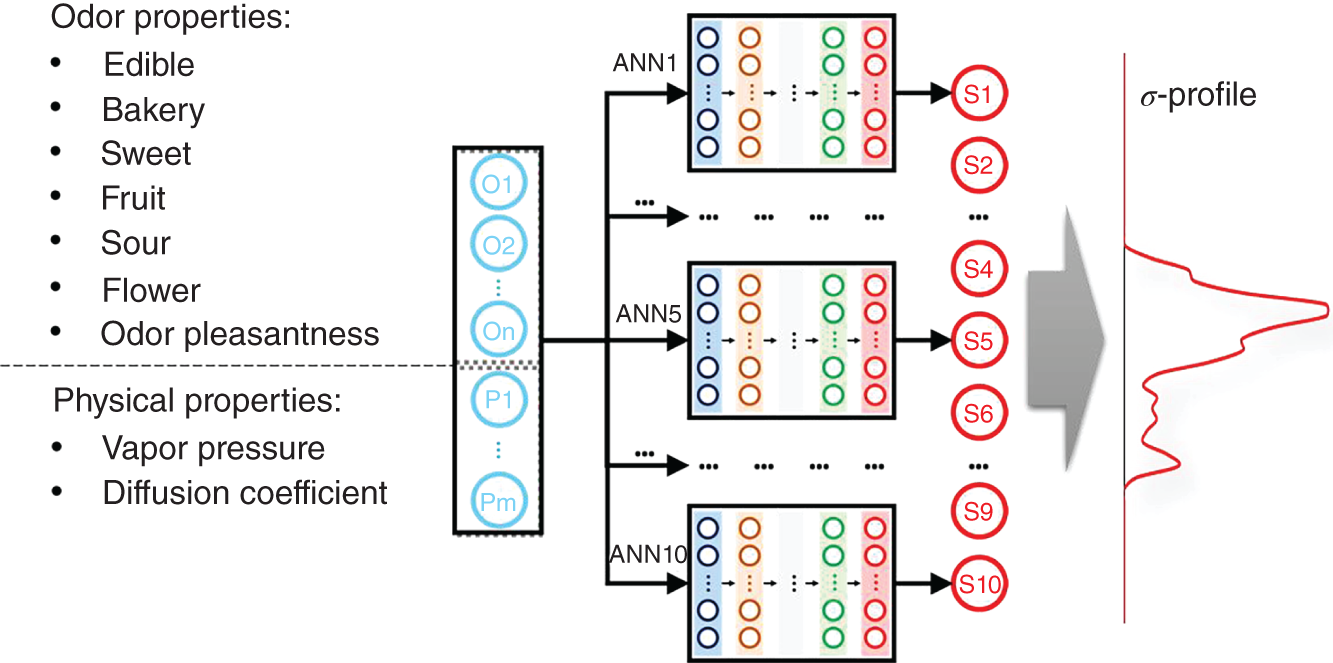
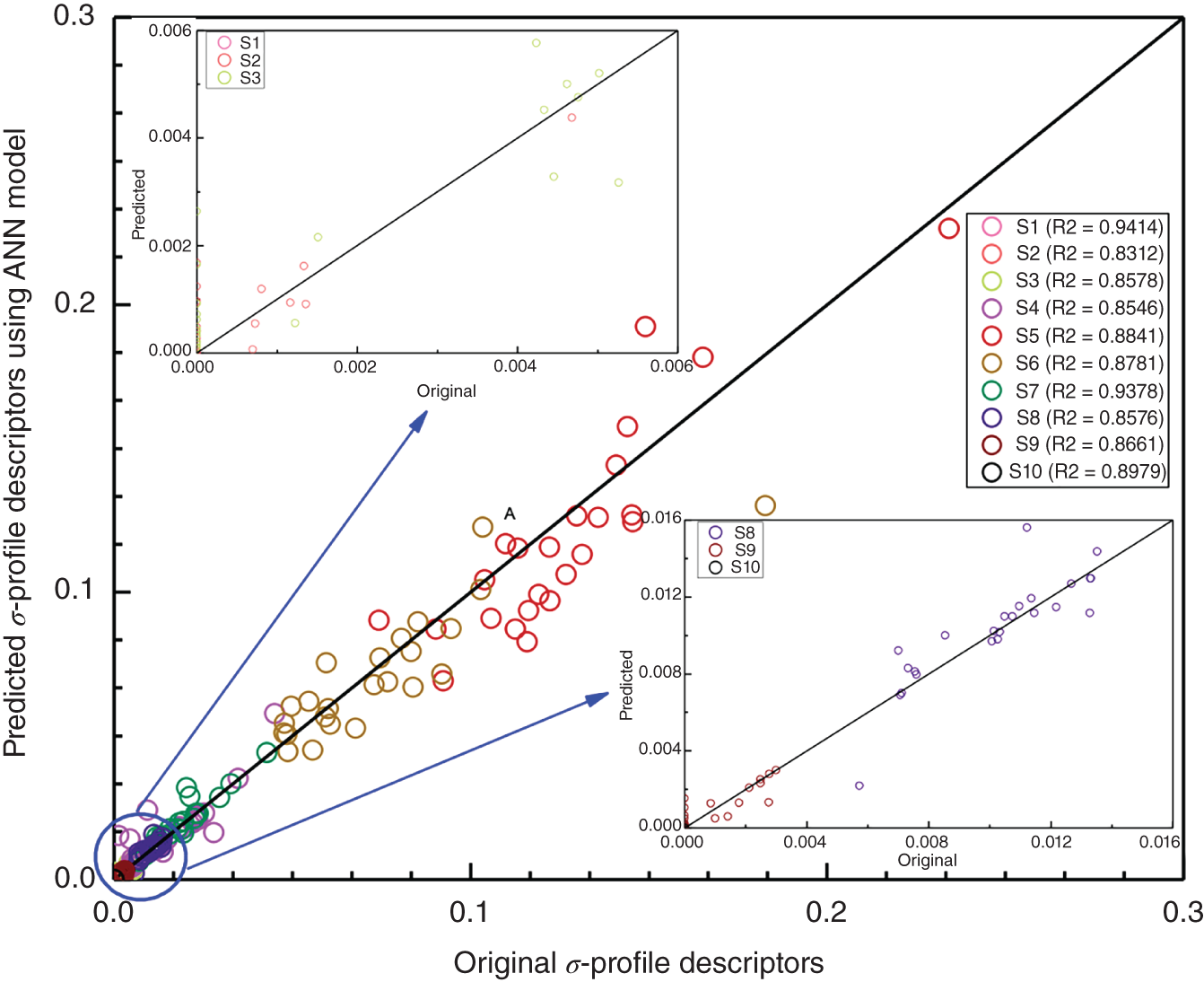
2.2.2.4 Results and Discussions
2.3 Computer-Aided Aroma Design (CAAD) Framework
2.3.1 CAAD for Pure Aromatic Chemicals
2.3.1.1 Identify Product Attributes

2.3.1.2 Convert Product Attributes to Properties and Their Constraints
2.3.1.3 Choose Property Prediction Model for Estimating Properties
2.3.1.4 Formulate MILP/MINLP Model
2.3.1.5 Solve the Model Using Decomposition-Based Algorithm
2.3.1.6 Verification
2.3.2 CAAD for Mixture Aromatic Chemicals

2.3.2.1 Identify Product Attributes
2.3.2.2 Convert Product Attributes into Properties and Corresponding Constraints
2.3.2.3 Establish Property Models
2.3.2.4 Ingredient Screening
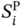 are the predicted values of σ-profiles area parameter i (i = 1, 2,…, 10) using the ANN-based SOR model.
are the predicted values of σ-profiles area parameter i (i = 1, 2,…, 10) using the ANN-based SOR model. 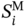 are the values of σ-profiles area parameter i of the designed aroma mixture.
are the values of σ-profiles area parameter i of the designed aroma mixture. 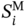 is calculated using Eq. (10).
is calculated using Eq. (10).
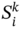 is the σ-profiles area parameter i of ingredient k, which is calculated using COSMO-SAC model and stored in the odor database.
is the σ-profiles area parameter i of ingredient k, which is calculated using COSMO-SAC model and stored in the odor database.
 , otherwise, xk = 0. In Eq. (2.12), n is the number of ingredients, and N is the upper bound of the ingredient number, which is set up by the users.
, otherwise, xk = 0. In Eq. (2.12), n is the number of ingredients, and N is the upper bound of the ingredient number, which is set up by the users.
2.3.2.5 Verification
2.4 Case Studies
2.4.1 Pure Aroma Design for Shampoo Additives
Properties
Constraints
Total group number
4 ≤ n ≤ 10
Repeat group number
ni ≤ 4
Functional group number
1 ≤ nF ≤ 3
Odor character
OC = sweet, fruit or flower
Odor pleasantness
OP ≥ 40
Diffusion coefficient (m2 h−1)
D ≥ 0.15
Vapor pressure (Pa)
Psat ≥ 100
Normal boiling point (K)
Tb ≥ 440
Normal melting point (K)
Tm ≤ 293.15
Solubility parameter (MPa1/2)
15 ≤ Sp ≤ 17
Viscosity (cp)
η ≤ 2
Density (g cm−3)
0.8 ≤ ρ ≤ 1
−log(LC50) (−log(mol l−1))
−log(LC50) ≤ 4.2 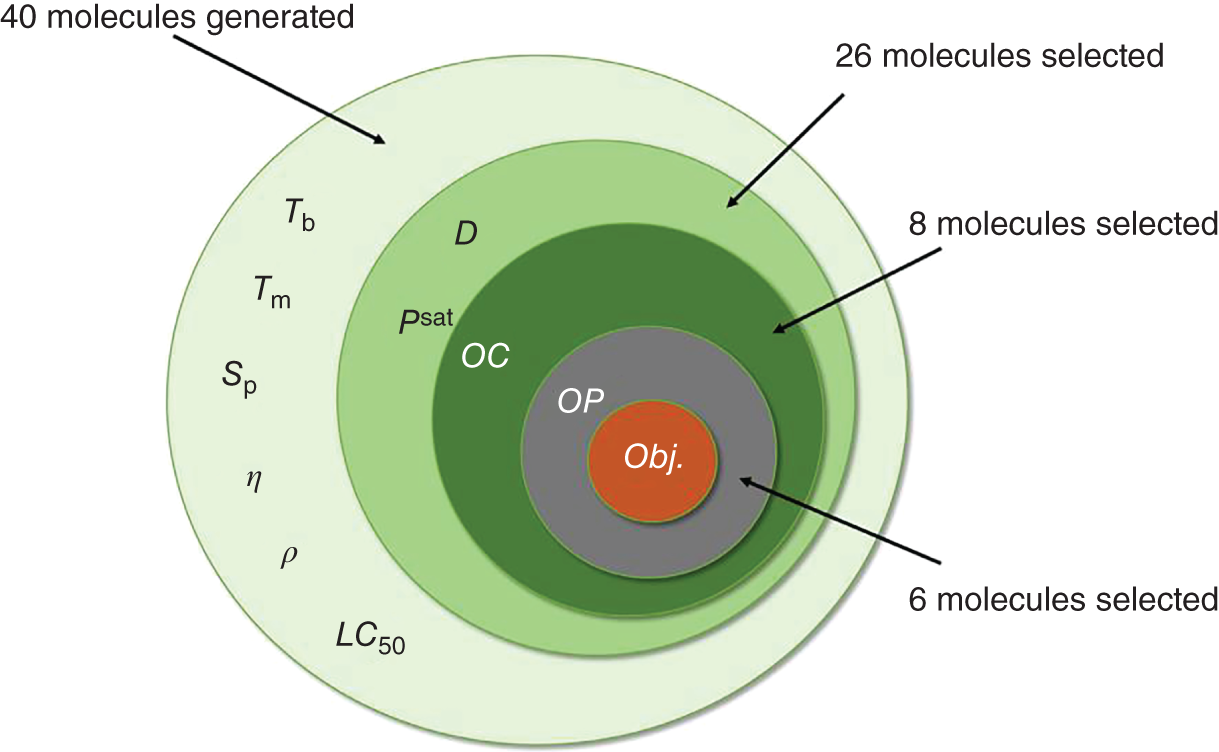
2.4.2 Pure Aroma Design for the Ingredient in Insect Repellent Spray
No.
1
2
3
4
5
6
Formula
C8H16O
C8H16O2
C7H12O2
C7H12O3
C8H14O2
C9H18O2
Groups
2 CH3
4 CH2
1 CH2CO
1 CH3
4 CH2
1 CH2CO
1 CH3O
1 CH3
1 CH
1 CH2=C
1 CH3CO
1 CH3O
1 CH3
1 CH
1 CH2=CH
1 CH2COO
1 CH3O
1 CH3
2 CH
1 CH2=CH
1 CH3CO
1 CH3O
3 CH3
3 CH2
1 C
1 CH3COO
Tm (K)
244
265
253
217
253
240
Tb (K)
443
469
443
442
459
458
Sp (Mpa1/2)
16.47
16.88
16.55
16.49
16.32
15.23
η (cp)
1.08
0.91
0.21
0.2
0.15
0.89
ρ (g cm−3)
0.82
0.9
0.96
1
0.94
0.9
−log(LC50)
3
2.58
2.83
3.53
3.58
3.13
Psat (Pa)
1003.8
138.2
838.4
1298.3
318.4
501.7
D (m2 h−1)
0.17
0.16
0.17
0.17
0.16
0.16
OC
Sweet
Sweet
Sweet
Sweet
Sweet
Sweet
OP
40
40
40
40
40
60
Available in database?
Y
Y
N
N
N
N
CAS No.
111-13-7
106-73-0
—
—
—
—
Molecular structure
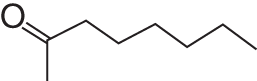
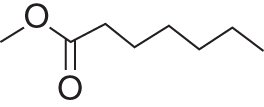
—
—
—
—
Odor in literature
Cheese-like, dairy nuances
Fruity
—
—
—
—
Chemical
wi (wt%)
Picaridin
9.74
2-Propanol
44.25
Water
42.01
Aroma
4
Properties
Constraints
Total group number
7 ≤ n ≤ 10
Repeat group number
ni ≤ 3
Functional group number
2 ≤ nF ≤ 3
Odor character
OC = sweet, fruit or flower
Odor pleasantness
OP ≥ 40
Diffusion coefficient (m2 h−1)
D ≥ 0.154
Vapor pressure (Pa)
Psat ≥ 14
Normal boiling point (K)
Tb ≥ 373.15
Normal melting point (K)
Tm ≤ 300
Solubility parameter (MPa1/2)
17 ≤ Sp ≤ 20
Viscosity (cp)
η ≤ 2
Molar volume (cm3 mol−1)
Vm ≤ 200
−log(LC50) (−log(mol l−1))
−log(LC50) ≤ 4.5
Molecular weight (g mol−1)
100 ≤ Mw ≤ 200
Flash point (K)
Fp ≥ 320
Phase stability
Solubility in the solvent mixture
2.4.3 Mixture Aroma Design for Aroma Substitutes
Odor property
Value
Physical property
Value
Ediblea)
33.42
Vapor pressure Psat (Pa)b)
53.86
Bakerya)
43.00
Diffusion coefficient DAB (m2 h−1)c)
0.16
Sweeta)
34.37
Solubility δ (mg l−1, 298 K)b)
158.9
Fruitya)
31.54
Boiling point Tb (K, 101.325 kPa)b)
454
Flowera)
29.95
Octanol–water partition coefficient log Ko/wb)
2.909
Soura)
25.93
Fathead minnow LC50 −log(LC50) (log mol l−1)c)
3.36
Pleasantnessa)
60.19
Flash point Fp (K)b)
333

Property
Constraint
δ (mg l−1, 298 K)
δ ≥ 158.6
Tb (K, 101.325 kPa)
Tb ≥ 333
Fp (K)
Fp ≥ 323
log Ko/w
log Ko/w ≤ 2.91
−log(LC50) (log mol l−1)
−log(LC50) ≤ 3.70 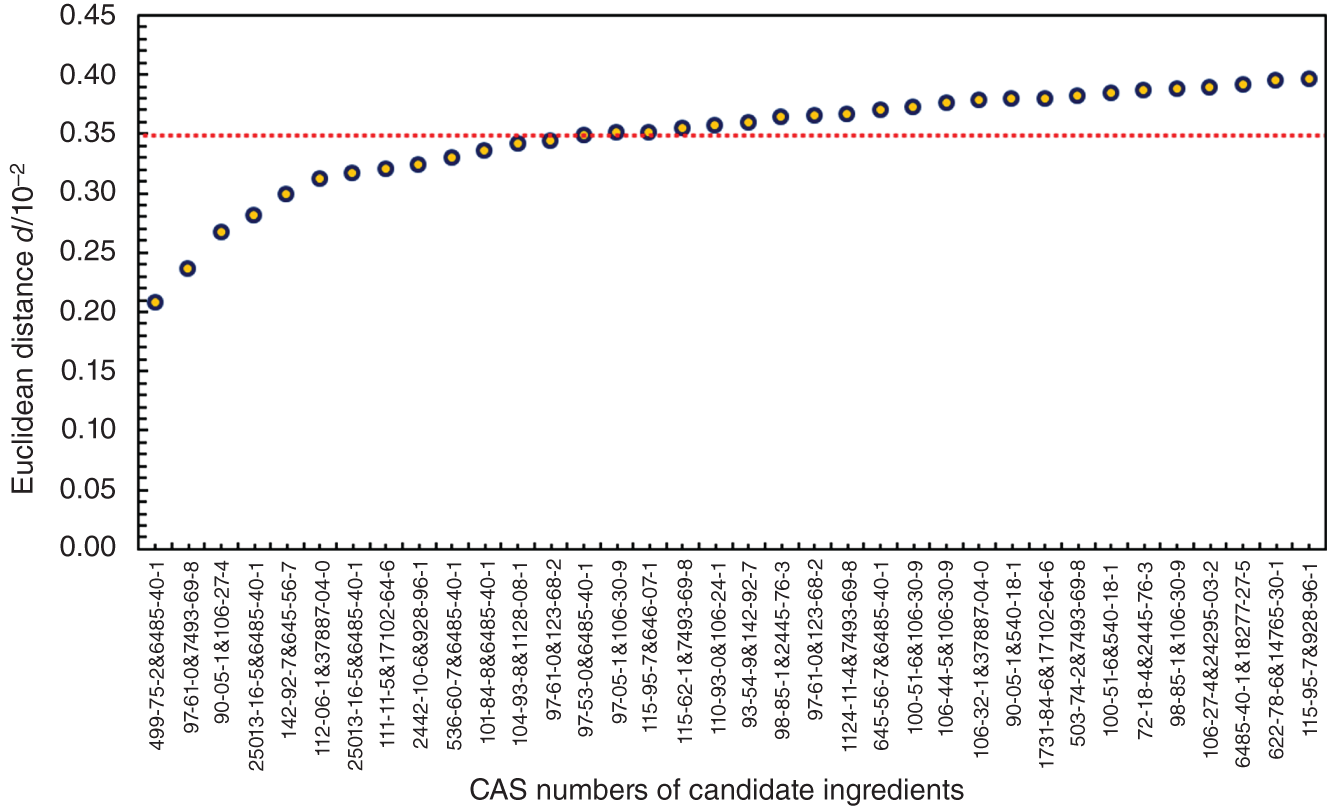
Designed aroma mixture
cis-3-Hexenyl propionate
Ingredient 1
Ingredient 2
CAS No.
33467-74-2 
97-61-0 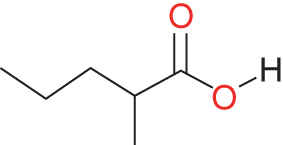
7493-69-8 
Odora)
Green, grass, flower, fruity
Sweet, edible, fruity
Fruity, edible
Volume fraction
1
0.4
0.6
Vapor pressure (Pa, 298 K)a)
53.86
23.99
85.46
Diffusion coefficientb)
0.156
0.172
0.156
Solubility (mg l−1, 298 K, water)a)
158.9
—
157.3
Boiling point (K, 101.325 kPa)a)
453–455
468.36
449.96
Flash point (K)a)
333
364.26
337.59
log Ko/wa)
2.909
1.8
2.972
−log(LC50) (log mol l−1)b)
3.36
2.45
4.03
Price (CNY/kg)c)
6,059
2,377
574
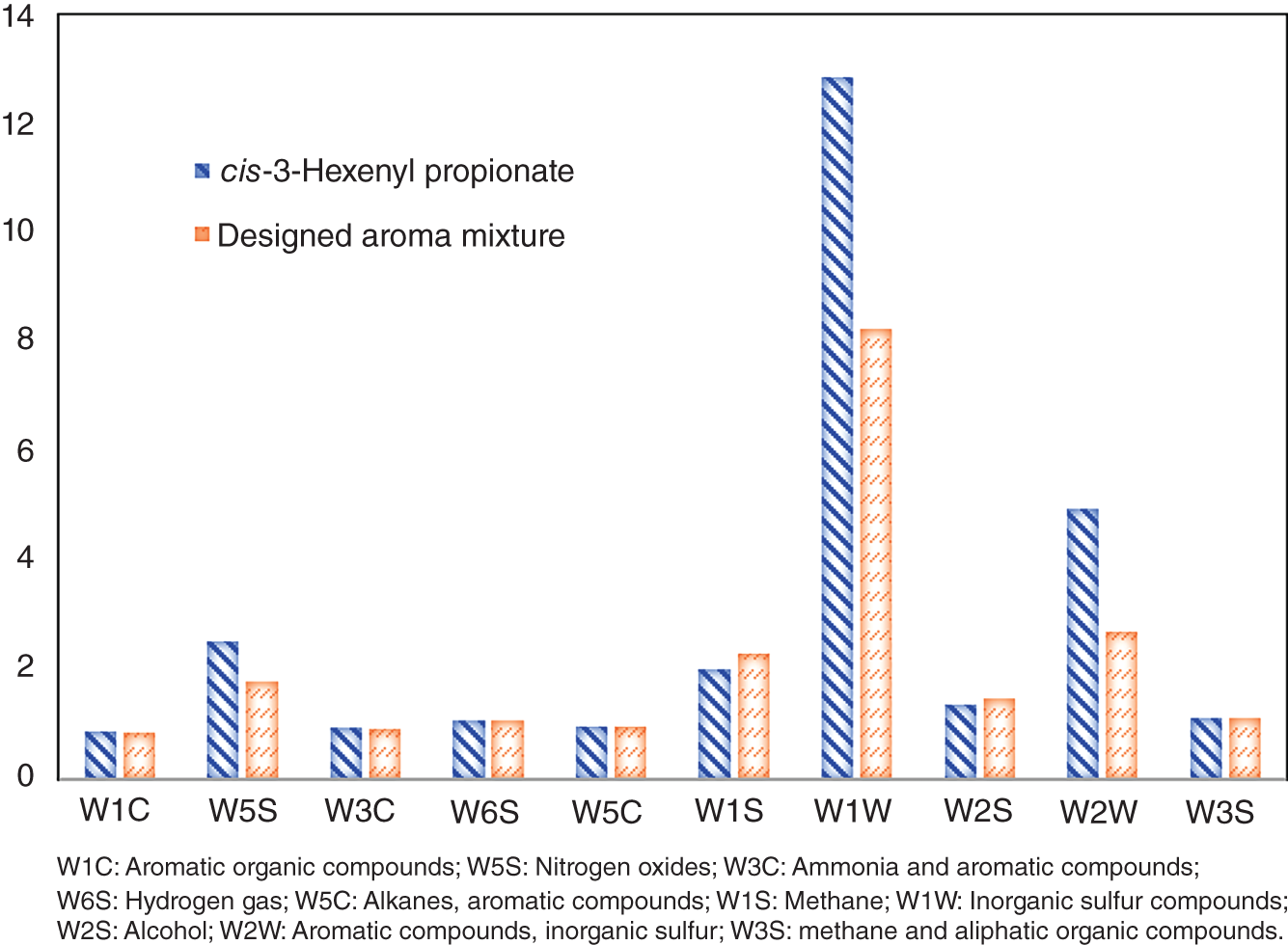
2.4.4 Mixture Aroma Design for Odor Tuning
Odor property
Value
Target value
Physical property
Value
Pleasantness
29.2
29.2
Vapor pressure Psat (Pa)a)
485.43
Edible
25.59
35.00
Diffusion coefficient DAB (m2 h−1)b)
0.174
Bakery
38.11
38.11
Solubility δ (mg l−1, 298 K)a)
1810
Sweet
12.46
30.00↑
Boiling point Tb (K, 101.325 kPa)a)
429.40
Fruit
11.55
25.00↑
Octanol–water partition coefficient log Ko/wa)
2.174
Sour
26.83
26.83
Fathead minnow LC50 −log(LC50) (log mol l−1)b)
3.68
Flower
24.33
24.33
Flash point Fp (K)a)
316.33
Designed aroma mixture
cis-4-Heptenal
Ingredient 1
Ingredient 2
CAS No.
6728-31-0
71-41-0
288-47-1
Volume fraction
1
0.9
0.1
Vapor pressure (Pa, 298 K)a)
485.43
293.31
2881.50
Diffusion coefficientb)
0.174
0.189
0.214
Solubility (mg l−1, 298 K, water)a)
1810
22000
53780
Boiling point (K, 101.325 kPa)a)
429.40
406.75
387.84
log Ko/wa)
2.174
1.51
0.44
−log(LC50) (log mol l−1)b)
3.68
2.83
2.94
Flash point (K)a)
316.33
321.89
295.22
Price (CNY/kg)c)
38 958.64
940.69
41179.3
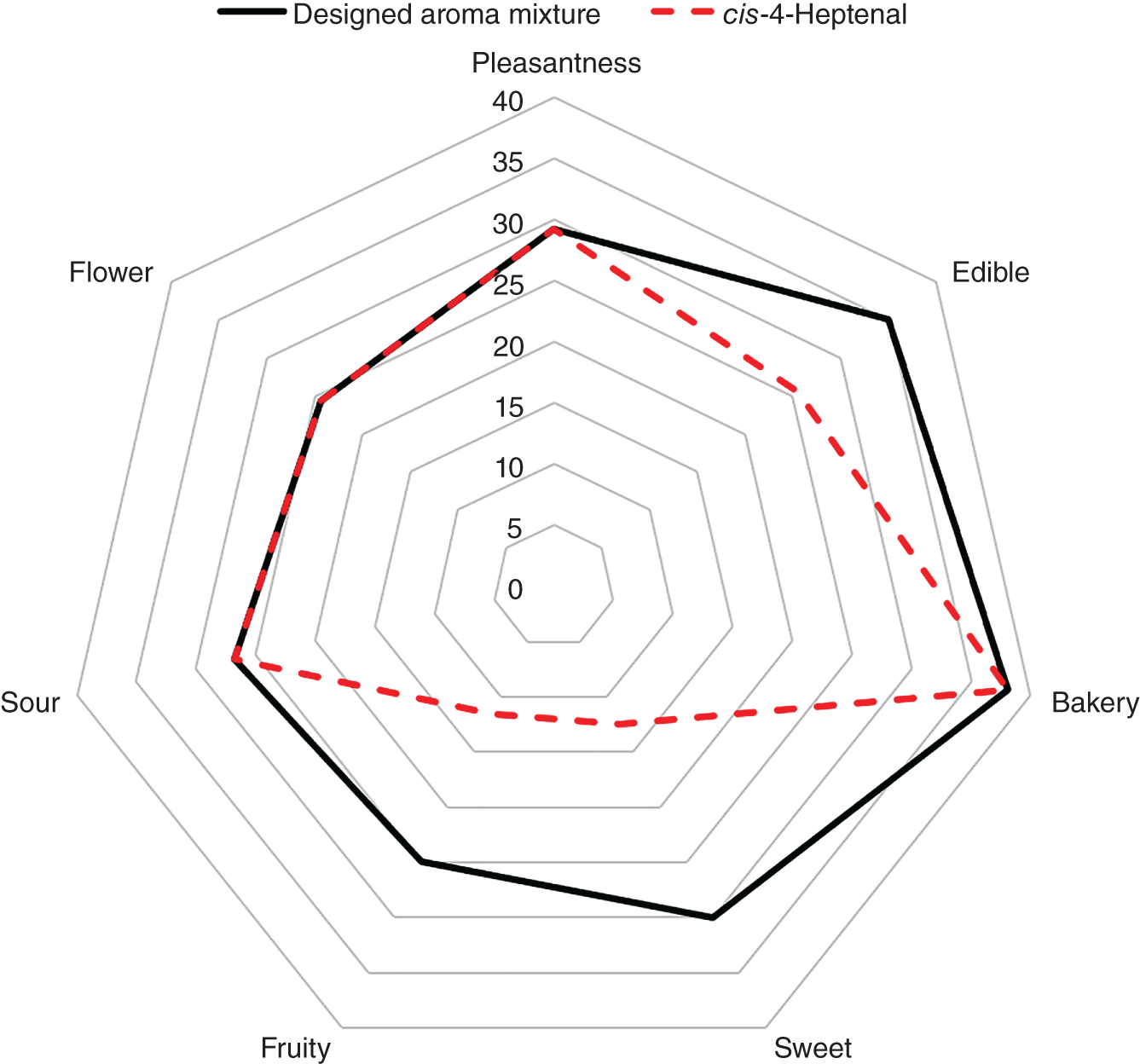
2.5 Conclusions
2.A The CAS Number of Molecules and the Selected Groups
100-06-1
111-14-8
13708-12-8
34413-35-9
592-88-1
7553-56-2
1003-04-9
111-27-3
137-32-6
34451-19-9
5989-27-5
7554-12-3
10031-82-0
111-47-7
13925-00-3
350-03-8
60-01-5
7558-79-4
100-41-4
111-61-5
140-10-3
35250-53-4
60047-17-8
75-65-0
100-51-6
111-65-9
140-11-4
352-93-2
60-12-8
76-22-2
100-52-7
111-66-0
140-39-6
35836-72-7
60-18-4
76-49-3
100-66-3
111-70-6
140-67-0
3658-80-8
6032-29-7
765-70-8
101-41-7
1117-55-1
141-12-8
36653-82-4
60-33-3
7732-18-5
101-84-8
111-79-5
141-78-6
3681-71-8
606-45-1
7764-50-3
102-04-5
111-80-8
142-19-8
37887-04-0
611-13-2
7779-41-1
102-13-6
111-87-5
142-62-1
3796-70-1
613-70-7
7779-50-2
102-16-9
1120-21-4
142-92-7
39212-23-2
614-99-3
7779-65-9
102-20-5
112-06-1
143-07-7
39255-32-8
616-25-1
77-83-8
102-76-1
112-12-9
143-08-8
40018-26-6
61-90-5
7785-70-8
103-05-9
1122-62-9
143-13-5
4075-07-4
61931-81-5
7786-58-5
103-09-3
112-30-1
1438-94-4
4077-47-8
620-02-0
7787-20-4
103-36-6
112-31-2
14765-30-1
4112-89-4
622-78-6
77-90-7
103-45-7
112-37-8
147-85-3
41453-56-9
623-37-0
77-92-9
103-50-4
1124-11-4
149-57-5
41519-23-7
623-42-7
77-93-0
103-60-6
112-42-5
150-30-1
4180-23-8
624-24-8
78-59-1
104-21-2
112-44-7
150-60-7
4208-49-5
625-84-3
78-70-6
104-54-1
1125-21-9
150-78-7
4221-99-2
626-93-7
78761-38-3
104-76-7
112-53-8
151-05-3
42436-07-7
627-90-7
78-83-1
104-93-8
1125-88-8
151-10-0
431-03-8
628-63-7
78-84-2
10519-33-2
112-66-3
1516-17-2
4411-89-6
629-19-6
78-93-3
10521-91-2
1128-08-1
15356-60-2
464-49-3
629-33-4
79-09-4
105-37-3
1131-62-0
15679-12-6
470-82-6
63012-97-5
79-20-9
105-53-3
115-95-7
15679-19-3
4864-61-3
6309-51-9
79-31-2
105-54-4
118-58-1
15707-23-0
491-35-0
63-68-3
79-77-6
105-57-7
118-93-4
15707-24-1
4938-52-7
637-64-9
80-62-6
105-60-2
1191-43-1
1618-26-4
498-02-2
6378-65-0
81-14-1
105-66-8
119-36-8
16409-46-4
499-75-2
638-25-5
81925-81-7
105-87-3
1193-79-9
16630-66-3
502-42-1
63-91-2
821-41-0
10599-70-9
119-61-9
1679-07-8
503-74-2
64-04-0
821-55-6
106-02-5
119-84-6
17102-64-6
505-10-2
64-17-5
823-22-3
106-21-8
120-14-9
1731-84-6
505-79-3
64-19-7
825-51-4
106-24-1
120-24-1
1759-28-0
50-69-1
64275-73-6
85-91-6
106-25-2
120-50-3
1797-74-6
50-70-4
645-56-7
87-22-9
106-27-4
120-51-4
18172-67-3
513-85-9
646-07-1
88-15-3
106-30-9
120-57-0
18277-27-5
5146-66-7
6485-40-1
881-68-5
106-32-1
120-58-1
18368-91-7
51-67-2
65405-77-8
882-33-7
106-36-5
120-92-3
18409-17-1
5271-38-5
656-53-1
88-69-7
106-44-5
121-32-4
18486-69-6
52-90-4
65-85-0
89-79-2
106-65-0
121-33-5
18640-74-9
529-20-4
66-25-1
89-83-8
106-68-3
121-34-6
1866-31-5
5292-21-7
67-03-8
90-00-6
106-70-7
122-03-2
2035-99-6
531-59-9
6728-26-3
90-02-8
106-73-0
122-40-7
2051-78-7
534-22-5
6728-31-0
90-05-1
1076-56-8
122-43-0
21145-77-7
536-60-7
67-56-1
91-22-5
107-85-7
122-63-4
2153-26-6
539-82-2
67-63-0
91-62-3
107-92-6
122-67-8
2257-09-2
540-07-8
67-64-1
91-64-5
107-95-9
122-70-3
2305-25-1
540-18-1
67-68-5
92-48-8
108-21-4
122-74-7
2396-84-1
540-42-1
67-71-0
92-52-4
108-22-5
122-97-4
24295-03-2
5405-41-4
67715-80-4
927-49-1
108-29-2
123-08-0
2432-51-1
541-31-1
67883-79-8
928-96-1
108-48-5
123-11-5
2442-10-6
54300-08-2
6789-88-4
93-04-9
108-94-1
123-19-3
2445-76-3
5461-08-5
6790-58-5
93-08-3
108-98-5
123-25-1
24683-00-9
5471-51-2
693-54-9
93-15-2
109-15-9
123-32-0
2497-18-9
54947-74-9
693-95-8
93-16-3
109-19-0
123-51-3
25013-16-5
551-93-9
698-10-2
93-28-7
109-21-7
123-66-0
25152-85-6
554-12-1
705-86-2
93-29-8
109-42-2
123-68-2
2530-10-1
56-40-6
706-14-9
93-51-6
109-52-4
123-72-8
2568-25-4
56-41-7
71-00-1
93-54-9
109-60-4
123-75-1
2623-23-6
56-81-5
710-04-3
93-58-3
109-73-9
123-86-4
2639-63-6
56-84-8
71-23-8
93-60-7
109-94-4
123-92-2
2705-87-5
56-85-9
71-36-3
93-89-0
110-02-1
124-04-9
2721-22-4
56-86-0
71-41-0
93905-03-4
110-15-6
124-06-1
2785-89-9
56-87-1
71-43-2
94278-27-0
110-17-8
124-07-2
27939-60-2
57074-37-0
7149-32-8
94-47-3
110-27-0
124-13-0
2847-30-5
57-55-6
7217-59-6
94-62-2
110-38-3
127-17-3
28664-35-9
583-60-8
72-18-4
96-22-0
110-40-7
128-37-0
288-47-1
584-02-1
7452-79-1
96-48-0
110-43-0
133-18-6
290-37-9
589-92-4
74-79-3
97-42-7
110-62-3
133-37-9
3149-28-8
589-98-0
7493-63-2
97-53-0
110-81-6
134-20-3
31704-80-0
590-01-2
7493-69-8
97-54-1
110-82-7
135-02-4
3208-16-0
59020-90-5
7493-71-2
97-61-0
110-86-1
13532-18-8
3268-49-3
591-24-2
75-07-0
97-99-4
110-89-4
13623-11-5
32974-92-8
591-31-1
75-18-3
98-00-0
110-93-0
13678-67-6
334-48-5
591-78-6
7540-51-4
98-85-1
111-11-5
137-00-8
33467-74-2
591-80-0
7541-49-3
98-86-2
111-13-7
137-06-4
3391-86-4
592-84-7
75-47-8
99-87-6
2.B The Calculation Formula of Odor Score
CH3
AC
CH3COO
CH2N
HCON(CH2)2
CH2
ACCH3
CH2COO
ACNH2
COO
CH
ACCH2
HCOO
C5H5N
CONH2
C
ACCH
CH3O
C5H4N
CONHCH2
CH2=CH
OH
CH2O
C5H3N
C2H4O2
CH=CH
CH3OH
CHO
COOH
CH3S
CH2=C
H2O
CH2NH2
CH2SH
CH2S
CH=C
ACOH
CHNH2
I
CHS
C=C
CH3CO
CH3NH
C#C
C4H4S
ACH
CH2CO
CH2NH
DMSO
C4H3S
2.C The Parameters and Results of the ANN Model
Model structure
Model parameters
Model output
R2 of training set
R2 of testing set
9-24-(Tanh)-20-(Tanh)-16-(Sigmoid)-12-(Tanh)-1
Weight initialization = Xavier;
Bias initialization = Zero;
Optimizer = Adam;
Epoch = 7000;
S1
0.9999
0.9905
9-24-(Tanh)-20-(Tanh)-16-(Sigmoid)-1
Weight initialization = Normal;
Bias initialization = Zero;
Optimizer = RMSprop;
Epoch = 6000;
S2
0.9783
0.9188
9-24-(Tanh)-20-(Sigmoid)-1
Weight initialization = Normal;
Bias initialization = Zero;
Optimizer = Levenberg–Marquardt;
Epoch = 4000;
S3
0.9850
0.9563
9-24-(Tanh)-20-(Sigmoid)-1
Weight initialization = Uniform;
Bias initialization = Zero;
Optimizer = Levenberg–Marquardt;
Epoch = 3000;
S4
0.9805
0.9272
9-18-(Tanh)-1
Weight initialization = Uniform;
Bias initialization = Zero;
Optimizer = Levenberg–Marquardt;
Epoch = 1000;
S5
0.9101
0.7764
9-18-(Tanh)-1
Weight initialization = Uniform;
Bias initialization = Zero;
Optimizer = Levenberg–Marquardt;
Epoch=1000;
S6
0.9007
0.8289
9-24-(Tanh)-20-(Tanh)-1
Weight initialization = Uniform;
Bias initialization = Zero;
Optimizer = Levenberg–Marquardt;
Epoch = 3000;
S7
0.9931
0.9018
9-24-(Tanh)-20-(Sigmoid)-1
Weight initialization = Normal;
Bias initialization = Zero;
Optimizer = Levenberg–Marquardt;
Epoch = 4000;
S8
0.9874
0.8735
9-24-(Tanh)-20-(Tanh)-16-(Sigmoid)-1
Weight initialization = Normal;
Bias initialization = Zero;
Optimizer = RMSprop;
Epoch = 6000;
S9
0.9773
0.8747
9-24-(Tanh)-20-(Tanh)-16-(Sigmoid)-12-(Tanh)-1
Weight initialization = Xavier;
Bias initialization = Zero;
Optimizer = Adam;
Epoch = 7000;
S10
0.9999
0.9998
2.D The Designed Results of Molecules for Case Study 2
No.
Groups
OC
OP
Available in database?
CAS number
Molecular structure
Odor in literature
1
3 CH3
1 CH
1 C
1 CH2=CH
1 OH
1 CH2COO
Sweet
40
N
—
—
—
2
3 CH3
2 CH2
1 C
1 OH
1 CH2COO
Sweet
40
N
—
—
—
3
2 CH3
1 CH2
1 C
1 CH2=CH
1 OH
1 CH2COO
Sweet
40
N
—
—
—
4
3 CH3
1 C
1 CH2=C
1 OH
1 CH2COO
Sweet
40
N
—
—
—
5
3 CH3
1 C
1 CH=CH
1 OH
1 CH3COO
Sweet
40
N
—
—
—
6
2 CH3
1 CH2
1 C
1 CH2=C
1 OH
1 CH3COO
Sweet
40
N
—
—
—
7
2 CH3
2 CH
1 CH2=CH
1 OH
1 CH2COO
Sweet
40
N
—
—
—
8
2 CH3
1 CH2
1 CH
1 CH2=C
1 OH
1 CH2COO
Sweet
40
N
—
—
—
9
3 CH3
2 CH2
1 C
1 CH2=CH
1 CH=C
1 OH
Sweet
40
Y
78-70-6
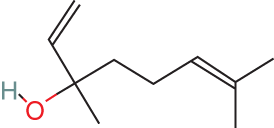
Sweet, floral, petitgrain-like
10
3 CH3
1 CH2
1 C
1 OH
1 CH2COO
Sweet
40
N
—
—
—
11
2 CH3
2 CH2
1 CH
1 OH
1 CH2COO
Sweet
40
N
—
—
—
12
3 CH3
2 CH
1 OH
1 CH2COO
Sweet
40
N
—
—
—
13
1 CH3
3 CH2
1 CH
1 OH
1 CH3COO
Sweet
40
N
—
—
—
14
3 CH3
3 CH2
1 CH
1 CH=C
1 OH
Sweet
40
N
—
—
—
15
2 CH3
2 CH2
1 CH
1 CH=CH
1 CH2=C
1 OH
Sweet
40
N
—
—
—
2.E Aroma Compounds for Ingredient Screening
CAS No.
Name
Smiles
100-06-1
Acetanisole
COc1ccc(cc1)C(C)=O
1003-04-9
45-Dihydro-3(2H)-thiophenone
O=C1CCSC1
10031-82-0
4-Ethoxybenzaldehyde
CCOc1ccc(C=O)cc1
100-41-4
Ethylbenzene
CCc1ccccc1
100-51-6
Benzyl alcohol
OCc1ccccc1
100-52-7
Benzaldehyde
O=Cc1ccccc1
100-66-3
Anisole
COc1ccccc1
101-41-7
Methyl phenylacetate
COC(=O)Cc1ccccc1
101-84-8
Diphenyl ether
O(c1ccccc1)c2ccccc2
102-04-5
13-Diphenyl-2-propanone
O=C(Cc1ccccc1)Cc2ccccc2
102-13-6
Isobutyl phenylacetate
CC(C)COC(=O)Cc1ccccc1
102-16-9
Benzyl phenylacetate
O=C(Cc1ccccc1)OCc2ccccc2
102-20-5
Phenethyl phenylacetate
O=C(Cc1ccccc1)OCCc2ccccc2
102-76-1
Triacetin
CC(=O)OCC(COC(C)=O)OC(C)=O
103-05-9
Dimethyl benzene propanol
CC(C)(O)CCc1ccccc1
103-09-3
2-Ethylhexyl acetate
CCCCC(CC)COC(C)=O
103-36-6
Ethyl cinnamate
CCOC(=O)\C=C\c1ccccc1
103-45-7
Phenethyl acetate
CC(=O)OCCc1ccccc1
103-50-4
Benzyl ether
C(OCc1ccccc1)c2ccccc2
103-60-6
2-Phenoxyethyl isobutyrate
CC(C)C(=O)OCCOc1ccccc1
104-21-2
Anisyl acetate
COc1ccc(COC(C)=O)cc1
104-54-1
Cinnamyl alcohol
OC\C=C/c1ccccc1
104-76-7
2-Ethyl-1-hexanol
CCCCC(CC)CO
104-93-8
4-Methylanisole
COc1ccc(C)cc1
10519-33-2
3-Decen-2-one
CCCCCC/C=C/C(C)=O
10521-91-2
5-Phenyl-1-pentanol
OCCCCCc1ccccc1
105-37-3
Ethyl propionate
CCOC(=O)CC
105-53-3
Diethyl malonate
CCOC(=O)CC(=O)OCC
105-54-4
Ethyl butyrate
CCCC(=O)OCC
105-57-7
Acetal
CCOC(C)OCC
105-60-2
ε-Caprolactam
O=C1CCCCCN1
105-66-8
Propyl butyrate
CCCOC(=O)CCC
105-87-3
Geranyl acetate
CC(=O)OC/C=C(C)/CCC=C(C)C
10599-70-9
3-Acetyl-25-dimethylfuran
CC(=O)c1cc(C)oc1C
106-02-5
Ω-Pentadecalactone
O=C1CCCCCCCCCCCCCCO1
106-21-8
37-Dimethyl-1-octanol
CC(C)CCCC(C)CCO
106-24-1
Geraniol
CC(C)=CCC\C(C)=C\CO
106-25-2
Nerol
CC(C)=CCC\C(C)=C/CO
106-27-4
Isoamyl butyrate
CCCC(=O)OCCC(C)C
106-30-9
Ethyl heptanoate
CCCCCCC(=O)OCC
106-32-1
Ethyl octanoate
CCCCCCCC(=O)OCC
106-36-5
Propyl propionate
CCCOC(=O)CC
106-44-5
p-Cresol
Cc1ccc(O)cc1
106-65-0
Dimethyl succinate
COC(=O)CCC(=O)OC
106-68-3
3-Octanone
CCCCCC(=O)CC
106-70-7
Methyl caproate
CCCCCC(=O)OC
106-73-0
Methyl heptanoate
CCCCCCC(=O)OC
1076-56-8
1-Methyl-3-methoxy-4-isopropylbenzene
COc1cc(C)ccc1C(C)C
107-85-7
Isoamylamine
NCCC(C)C
107-92-6
Butyric acid
CCCC(O)=O
107-95-9
β-Alanine
NCCC(O)=O
108-21-4
Isopropyl acetate (replicate)
CC(C)OC(C)=O
108-22-5
Isopropenyl acetate
CC(=C)OC(C)=O
108-29-2
γ-Valerolactone
CC1CCC(=O)O1
108-48-5
26-Dimethylpyridine
Cc1cccc(C)n1
108-94-1
Cyclohexanone
O=C1CCCCC1
108-98-5
Benzenethiol
Sc1ccccc1
109-15-9
Octyl isobutyrate
CCCCCCCCOC(=O)C(C)C
109-19-0
Isobutyl acetate
O=C(C)OCC(C)C
109-21-7
Butyl butyrate
CCCCOC(=O)CCC
109-42-2
Butyl 10-undecenoate
CCCCOC(=O)CCCCCCCCC=C
109-52-4
Valeric acid
CCCCC(O)=O
109-60-4
Propyl acetate
CCCOC(C)=O
109-73-9
Butylamine
CCCCN
109-94-4
Ethyl formate
CCOC=O
110-02-1
Thiophene (replicate)
s1cccc1
110-15-6
Succinic acid
OC(=O)CCC(O)=O
110-17-8
Fumaric acid
OC(=O)\C=C\C(O)=O
110-27-0
Isopropyl myristate
CCCCCCCCCCCCCC(=O)OC(C)C
110-38-3
Ethyl decanoate
CCCCCCCCCC(=O)OCC
110-40-7
Diethyl sebacate
CCOC(=O)CCCCCCCCC(=O)OCC
110-43-0
2-Heptanone
CCCCCC(C)=O
110-62-3
Valeraldehyde
CCCCC=O
110-81-6
Diethyl disulfide
CCSSCC
110-82-7
Cyclohexane
C1CCCCC1
110-86-1
Pyridine
c1ccncc1
110-89-4
Piperidine
C1CCNCC1
110-93-0
6-Methyl-5-hepten-2-one
CC(C)=CCCC(C)=O
111-11-5
Methyl caprylate
CCCCCCCC(=O)OC
111-13-7
2-Octanone
CCCCCCC(C)=O
111-14-8
Heptanoic acid
CCCCCCC(O)=O
111-27-3
Hexanol
CCCCCCO
111-47-7
Propyl sulfide
CCCSCCC
111-61-5
Ethyl stearate
CCCCCCCCCCCCCCCCCC(=O)OCC
111-65-9
Octane
CCCCCCCC
111-66-0
1-Octene
CCCCCCC=C
111-70-6
Heptanol
CCCCCCCO
1117-55-1
Hexyl octanoate
CCCCCCCC(=O)OCCCCCC
111-79-5
Methyl trans-2-nonenoate
CCCCCC\C=C/C(=O)OC
111-80-8
Methyl 2-nonynoate
CCCCCCC#CC(=O)OC
111-87-5
Octanol
CCCCCCCCO
1120-21-4
Undecane
CCCCCCCCCCC
112-06-1
Heptyl acetate
CCCCCCCOC(C)=O
112-12-9
2-Undecanone
CCCCCCCCCC(C)=O
1122-62-9
2-Acetyl pyridine
CC(=O)c1ccccn1
112-30-1
Decanol
CCCCCCCCCCO
112-31-2
Decanal
CCCCCCCCCC=O
112-37-8
Undecanoic acid
CCCCCCCCCCC(O)=O
1124-11-4
2356-Tetramethylpyrazine
Cc1nc(C)c(C)nc1C
112-42-5
1-Undecanol
CCCCCCCCCCCO
112-44-7
Undecanal
CCCCCCCCCCC=O
1125-21-9
4-Oxoisophorone
CC1=CC(=O)CC(C)(C)C1=O
112-53-8
Lauryl alcohol
CCCCCCCCCCCCO
1125-88-8
Benzaldehyde dimethyl acetal
COC(OC)c1ccccc1
112-66-3
Lauryl acetate
CCCCCCCCCCCCOC(C)=O
1128-08-1
Dihydrojasmone
CCCCCC1=C(C)CCC1=O
1131-62-0
34-Dimethoxyacetophenone
COc1ccc(cc1OC)C(C)=O
115-95-7
Linalyl acetate
CC(C)=CCCC(C)(OC(C)=O)C=C
118-58-1
Benzyl salicylate
Oc1ccccc1C(=O)OCc2ccccc2
118-93-4
2-Hydroxyacetophenone
CC(=O)c1ccccc1O
1191-43-1
16-Hexanedithiol
SCCCCCCS
119-36-8
Methyl salicylate
COC(=O)c1ccccc1O
1193-79-9
2-Acetyl-5-methylfuran
CC(=O)c1oc(C)cc1
119-61-9
Benzophenone
O=C(c1ccccc1)c2ccccc2
119-84-6
Dihydrocoumarin
O=C1CCc2ccccc2O1
120-14-9
Veratraldehyde
COc1ccc(C=O)cc1OC
120-24-1
Isoeugenyl phenylacetate
COc1cc(\C=C\C)ccc1OC(=O)Cc2ccccc2
120-50-3
Isobutyl benzoate
CC(C)COC(=O)c1ccccc1
120-51-4
Benzyl benzoate
O=C(OCc1ccccc1)c2ccccc2
120-57-0
Piperonal
O=Cc1ccc2OCOc2c1
120-58-1
Isosafrole
C/C=C/c1ccc2OCOc2c1
120-92-3
Cyclopentanone
O=C1CCCC1
121-32-4
Ethyl Vanillin
CCOc1cc(C=O)ccc1O
121-33-5
Vanillin
COc1cc(C=O)ccc1O
121-34-6
4-Hydroxy-3-methoxybenzoic acid
COc1cc(ccc1O)C(O)=O
122-03-2
Cuminaldehyde
CC(C)c1ccc(C=O)cc1
122-40-7
α-Amylcinnamaldehyde
CCCCC/C(C=O)=C/c1ccccc1
122-43-0
Butyl phenylacetate
CCCCOC(=O)Cc1ccccc1
122-63-4
Benzyl propionate
CCC(=O)OCc1ccccc1
122-67-8
Isobutyl cinnamate
CC(C)COC(=O)\C=C\c1ccccc1
122-70-3
Phenethyl propionate
CCC(=O)OCCc1ccccc1
122-74-7
3-Phenylpropyl propionate
CCC(=O)OCCCc1ccccc1
122-97-4
3-Phenyl-1-propanol
OCCCc1ccccc1
123-08-0
4-Hydroxybenzaldehyde
Oc1ccc(C=O)cc1
123-11-5
p-Anisaldehyde
COc1ccc(C=O)cc1
123-19-3
4-Heptanone
CCCC(=O)CCC
123-25-1
Diethyl succinate
CCOC(=O)CCC(=O)OCC
123-32-0
25-Dimethyl pyrazine
Cc1cnc(C)cn1
123-51-3
Isoamyl alcohol
CC(C)CCO
123-66-0
Ethyl hexanoate
CCCCCC(=O)OCC
123-68-2
Allyl hexanoate
CCCCCC(=O)OCC=C
123-72-8
Butyraldehyde
CCCC=O
123-75-1
Pyrrolidine
N1CCCC1
123-86-4
Butyl acetate
CCCCOC(C)=O
123-92-2
Isopentyl acetate
CC(C)CCOC(C)=O
124-04-9
Adipic acid
OC(=O)CCCCC(O)=O
124-06-1
Ethyl myristate
CCCCCCCCCCCCCC(=O)OCC
124-07-2
Octanoic acid
CCCCCCCC(O)=O
124-13-0
Octanal
CCCCCCCC=O
127-17-3
Pyruvic acid
CC(=O)C(O)=O
128-37-0
Butylated hydroxytoluene
Cc1cc(c(O)c(c1)C(C)(C)C)C(C)(C)C
133-18-6
Phenethyl anthranilate
Nc1ccccc1C(=O)OCCc2ccccc2
133-37-9
DL-Tartaric acid
OC(C(O)C(O)=O)C(O)=O
134-20-3
Methyl anthranilate
COC(=O)c1ccccc1N
135-02-4
o-Anisaldehyde
COc1ccccc1C=O
13532-18-8
Methyl 3-(methylthio)propionate
COC(=O)CCSC
13623-11-5
245-Trimethylthiazole
Cc1sc(C)c(C)n1
13678-67-6
22′-(Thiodimethylene)difuran
C(SCc1occc1)c2occc2
137-00-8
4-Methyl-5-thiazoleethanol
Cc1ncsc1CCO
137-06-4
o-Toluenethiol
Cc1ccccc1S
13708-12-8
5-Methylquinoxaline
Cc1cccc2nccnc12
137-32-6
2-Methyl-1-butanol (replicate)
CCC(C)CO
13925-00-3
2-Ethyl pyrazine
CCc1cnccn1
140-10-3
trans-Cinnamic acid
OC(=O)\C=C\c1ccccc1
140-11-4
Benzyl acetate
CC(=O)OCc1ccccc1
140-39-6
p-Tolyl acetate
CC(=O)Oc1ccc(C)cc1
140-67-0
4-Allylanisole
COc1ccc(CC=C)cc1
141-12-8
Neryl acetate
CC(=O)OC\C=C(C)/CCC=C(C)C
141-78-6
Ethyl acetate
CCOC(C)=O
142-19-8
Allyl heptanoate
CCCCCCC(=O)OCC=C
142-62-1
Caproic acid
CCCCCC(O)=O
142-92-7
Hexyl acetate
CCCCCCOC(C)=O
143-07-7
Lauric acid (replicate)
CCCCCCCCCCCC(O)=O
143-08-8
Nonanol
CCCCCCCCCO
143-13-5
Nonyl acetate
CCCCCCCCCOC(C)=O
1438-94-4
1-Furfurylpyrrole
C(n1cccc1)c2occc2
14765-30-1
2-sec-Butylcyclohexanone
CCC(C)C1CCCCC1=O
147-85-3
L-Proline
OC(=O)C1CCCN1
149-57-5
2-Ethylhexanoic acid
CCCCC(CC)C(O)=O
150-30-1
DL-Phenylalanine (replicate)
NC(Cc1ccccc1)C(O)=O
150-60-7
Benzyl disulfide
C(SSCc1ccccc1)c2ccccc2
150-78-7
Dimethoxy benzene
COc1ccc(OC)cc1
151-05-3
α,α-Dimethylphenethyl acetate
CC(=O)OC(C)(C)Cc1ccccc1
151-10-0
13-Dimethoxybenzene
COc1cccc(OC)c1
1516-17-2
transtrans-24-Hexadienyl acetate (replicate)
C/C=C/C=C/COC(C)=O
15356-60-2
(+)-Menthol
CC(C)[C@@H]1CC[C@@H](C)C[C@@H]1O
15679-12-6
2-Ethyl-4-methylthiazole
CCc1scc(C)n1
15679-19-3
2-Ethoxythiazole
CCOc1sccn1
15707-23-0
2-Ethyl-3-methylpyrazine
CCc1nccnc1C
15707-24-1
23-Diethylpyrazine
CCc1nccnc1CC
1618-26-4
Bis(methylthio)methane
CSCSC
16409-46-4
Menthyl isovalerate
CCC(C)C(=O)OC1CC(C)CCC1C(C)C
16630-66-3
Methyl (methylthio)acetate
COC(=O)CSC
1679-07-8
Cyclopentanethiol
SC1CCCC1
17102-64-6
transtrans-24-Hexadien-1-ol
C/C=C/C=C/CO
1731-84-6
Methyl nonanoate
CCCCCCCCC(=O)OC
1759-28-0
4-Methyl-5-vinylthiazole
Cc1ncsc1C=C
1797-74-6
Allyl phenylacetate
C=CCOC(=O)Cc1ccccc1
18172-67-3
(−)-β-Pinene
CC1(C)[C@H]2CCC(=C)[C@@H]1C2
18277-27-5
2-(1-Methylpropyl)thiazole
CCC(C)c1sccn1
18368-91-7
2-Ethylfenchol
CCC1(O)C(C)(C)C2CCC1(C)C2
18409-17-1
trans-2-Octen-1-ol
CCCCC\C=C\CO
18486-69-6
(1R)-(−)-Myrtenal
O=C(C)C1=CCC2CC1C2(C)C
18640-74-9
2-Isobutylthiazole
CC(C)Cc1sccn1
1866-31-5
Allyl cinnamate
C=CCOC(=O)\C=C\c1ccccc1
2035-99-6
Isoamyl octanoate
CCCCCCCC(=O)OCCC(C)C
2051-78-7
Allyl butyrate
CCCC(=O)OCC=C
21145-77-7
6-Acetyl-112447-Hexamethyltetralin
CC1CC(C)(C)c2cc(C(C)=O)c(C)cc2C1(C)C
2153-26-6
Terpinyl formate
CC1=CCC(CC1)C(C)(C)OC=O
2257-09-2
2-Phenylethyl isothiocyanate
S=C=NCCc1ccccc1
2305-25-1
Ethyl-3-hydroxyhexanoate
CCCC(O)CC(=O)OCC
2396-84-1
Ethyl sorbate
CCOC(=O)\C=C\C=C\C
24295-03-2
2-Acetylthiazole
CC(=O)c1sccn1
2432-51-1
Methyl thiobutyrate
CCCC(=O)SC
2442-10-6
1-Octen-3-yl acetate
CCCCCC(OC(C)=O)C=C
2445-76-3
Hexyl propionate
CCCCCCOC(=O)CC
24683-00-9
2-Isobutyl-3-methoxypyrazine
COc1nccnc1CC(C)C
2497-18-9
trans-2-Hexenyl acetate
CCC\C=C/COC(C)=O
25013-16-5
Butylated hydroxyanisole
COc1ccc(O)c(c1)C(C)(C)C
25152-85-6
cis-3-Hexenyl benzoate
CC\C=C/CCOC(=O)c1ccccc1
2530-10-1
3-Acetyl-25-dimethylthiophene
CC(=O)c1cc(C)sc1C
2568-25-4
Benzaldehyde propylene glycol acetal
CC1COC(O1)c2ccccc2
2623-23-6
L-Menthyl acetate (replicate)
CC(C)[C@@H]1CC[C@@H](C)C[C@H]1OC(C)=O
2639-63-6
Hexyl butyrate
CCCCCCOC(=O)CCC
2705-87-5
Allyl cyclohexanepropionate
C=CCOC(=O)CCC1CCCCC1
2721-22-4
δ-Tetradecalactone (replicate)
CCCCCCCCCC1CCCC(=O)O1
2785-89-9
4-Ethylguaiacol
CCc1ccc(O)c(OC)c1
27939-60-2
Trivertal
CC1C=CCCC1(C)C=O
2847-30-5
2-Methoxy-3-methylpyrazine
COc1nccnc1C
28664-35-9
45-Dimethyl-3-hydroxy-25-dihydrofuran-2-one
CC1OC(=O)C(=C1C)O
288-47-1
Thiazole
s1ccnc1
290-37-9
Pyrazine
c1cnccn1
3149-28-8
2-Methoxypyrazine
COc1cnccn1
31704-80-0
3-(5-Methyl-2-furyl)butanal
CCC(C=O)c1oc(C)cc1
3208-16-0
2-Ethylfuran
CCc1occc1
3268-49-3
3-(Methylthio)propionaldehyde
CSCCC=O
32974-92-8
2-Acetyl-3-ethylpyrazine
CCc1nccnc1C(C)=O
334-48-5
Decanoic acid
CCCCCCCCCC(O)=O
33467-74-2
cis-3-Hexenyl propionate
CC\C=C/CCOC(=O)CC
3391-86-4
1-Octen-3-ol
CCCCCC(O)C=C
34413-35-9
5678-Tetrahydroquinoxaline
C1CCc2nccnc2C1
34451-19-9
Butyl (S)-(−)-lactate
CCCCOC(=O)C(C)O
350-03-8
3-Acetylpyridine
CC(=O)c1cccnc1
35250-53-4
Pyrazineethanethiol
SCCc1cnccn1
352-93-2
Diethyl sulfide
CCSCC
35836-72-7
Nopol acetate
O=C(C)OCCC1CCC2CC1C2(C)C
3658-80-8
Dimethyl trisulfide (replicate)
CSSSC
36653-82-4
1-Hexadecanol
CCCCCCCCCCCCCCCCO
3681-71-8
cis-3-Hexenyl acetate
CC\C=C/CCOC(C)=O
37887-04-0
2-Mercapto-3-butanol
CC(O)C(C)S
3796-70-1
610-Dimethyl-59-undecadien-2-one
CC(C)=CCC\C(C)=C\CCC(C)=O
39212-23-2
Whiskey lactone
CCCCC1OC(=O)CC1C
39255-32-8
Ethyl 2-methylpentanoate (replicate)
CCCC(C)C(=O)OCC
40018-26-6
25-Dihydroxy-14-dithiane
OC1CSC(O)CS1
4075-07-4
Androstadienone
CC12CCC3C(CCC4=CC(=O)CCC34C)C1CC=C2
4077-47-8
25-Dimethyl-4-methoxy-3(2H)-furanone
COC1=C(C)OC(C)C1=O
4112-89-4
Guaiacyl phenylacetate
COc1ccccc1OC(=O)Cc2ccccc2
41453-56-9
cis-2-Nonen-1-ol
CCCCCC\C=C/CO
41519-23-7
cis-3-Hexenyl isobutyrate
CC\C=C/CCOC(=O)C(C)C
4180-23-8
trans-Anethole
COc1ccc(\C=C\C)cc1
4208-49-5
Allyl 2-furoate
C=CCOC(=O)c1occc1
4221-99-2
(S)-(+)-2-butanol
CCC(C)O
42436-07-7
cis-3-Hexenyl phenylacetate
CC\C=C/CCOC(=O)Cc1ccccc1
431-03-8
23-Butanedione
CC(=O)C(C)=O
4411-89-6
2-Phenyl-2-butenal
C\C=C(C=O)/c1ccccc1
464-49-3
D-Camphor
CC1(C)[C@H]2CC[C@@]1(C)C(=O)C2
470-82-6
18-Cineole
CC12CCC(CC1)C(C)(C)O2
4864-61-3
3-Octyl acetate
CCCCCC(CC)OC(=O)C
491-35-0
4-Methylquinoline
Cc1ccnc2ccccc12
4938-52-7
1-Hepten-3-ol
CCCCC(O)C=C
498-02-2
Acetovanillone
COc1cc(ccc1O)C(C)=O
499-75-2
Carvacrol
CC(C)c1ccc(C)c(O)c1
502-42-1
Cycloheptanone
O=C1CCCCCC1
503-74-2
Isovaleric acid
CC(C)CC(O)=O
505-10-2
3-(Methylthio)-1-propanol
CSCCCO
505-79-3
3-(Methylthio)propyl isothiocyanate
CSCCCN=C=S
50-69-1
D-(−)-Ribose
OCC1OC(O)C(O)C1O
50-70-4
D-Sorbitol
OCC(O)C(O)C(O)C(O)CO
513-85-9
Butanediol
C[C@@H](O)[C@H](C)O
5146-66-7
37-Dimethyl-26-octadienenitrile
CC(C)=CCC\C(C)=C\C#N
51-67-2
Tyramine
NCCc1ccc(O)cc1
5271-38-5
2-(Methylthio)ethanol
CSCCO
52-90-4
L-Cysteine
NC(CS)C(O)=O
529-20-4
o-Tolualdehyde
Cc1ccccc1C=O
5292-21-7
Cyclohexaneacetic acid
OC(=O)CC1CCCCC1
531-59-9
7-Methoxycoumarin
COc1ccc2C=CC(=O)Oc2c1
534-22-5
2-Methylfuran
Cc1occc1
536-60-7
4-Isopropylbenzyl alcohol
CC(C)c1ccc(CO)cc1
539-82-2
Ethyl valerate
CCCCC(=O)OCC
540-07-8
Amyl hexanoate
CCCCCOC(=O)CCCCC
540-18-1
Amyl butyrate
CCCCCOC(=O)CCC
540-42-1
Isobutyl propionate
CCC(=O)OCC(C)C
5405-41-4
Ethyl-3-hydroxybutyrate
CCOC(=O)CC(C)O
541-31-1
3-Methyl-1-butanethiol
CC(C)CCS
54300-08-2
2-Acetyl-35(6)-dimethylpyrazine
CC(=O)c1ncc(C)nc1C
5461-08-5
Piperonyl isobutyrate
CC(C)C(=O)OCc1ccc2OCOc2c1
5471-51-2
4-(4-Hydroxyphenyl)-2-butanone
CC(=O)CCc1ccc(O)cc1
54947-74-9
(±)-4-Methyloctanoic acid
CCCC[C@@H](C)CCC([O-])=O
551-93-9
2-Aminoacetophenone
CC(=O)c1ccccc1N
554-12-1
Methyl propionate
CCC(=O)OC
56-40-6
Glycine
NCC(O)=O
56-41-7
L-Alanine
CC(N)C(O)=O
56-81-5
Glycerol
OCC(O)CO
56-84-8
Aspartic acid
NC(CC(O)=O)C(O)=O
56-85-9
L-Glutamine
N[C@@H](CCC(N)=O)C(O)=O
56-86-0
L-Glutamic acid
NC(CCC(O)=O)C(O)=O
56-87-1
L-Lysine
NCCCCC(N)C(O)=O
57074-37-0
cis-4-Decen-1-ol
CCCCC\C=C\CCCO
57-55-6
Propylene glycol
CC(O)CO
583-60-8
2-Methylcyclohexanone
CC1CCCCC1=O
584-02-1
3-Pentanol
CCC(O)CC
589-92-4
4-Methylcyclohexanone
CC1CCC(=O)CC1
589-98-0
3-Octanol
CCCCCC(O)CC
590-01-2
Butyl propionate
CCCCOC(=O)CC
59020-90-5
2-Furanmethanethiol formate
O=CSCc1occc1
591-24-2
3-Methylcyclohexanone
CC1CCCC(=O)C1
591-31-1
m-Anisaldehyde
COc1cccc(C=O)c1
591-78-6
2-Hexanone
CCCCC(C)=O
591-80-0
4-Pentenoic acid
OC(=O)CCC=C
592-84-7
Butyl formate
CCCCOC=O
592-88-1
Allyl sulfide
C=CCSCC=C
5989-27-5
D-Limonene
CC(=C)[C@@H]1CCC(=CC1)C
60-01-5
Tributyrin
CCCC(=O)OCC(COC(=O)CCC)OC(=O)CCC
60047-17-8
Linalool oxide
CC(C)(O)C1CCC(C)(O1)C=C
60-12-8
2-Phenylethanol
OCCc1ccccc1
60-18-4
L-Tyrosine
NC(Cc1ccc(O)cc1)C(O)=O
6032-29-7
2-Pentanol
CCCC(C)O
60-33-3
Linoleic acid
CCCCC/C=C\C\C=C/CCCCCCCC(O)=O
606-45-1
Methyl 2-methoxybenzoate
COC(=O)c1ccccc1OC
611-13-2
Methyl 2-furoate
COC(=O)c1occc1
613-70-7
2-Methoxyphenyl acetate
COc1ccccc1OC(C)=O
614-99-3
Ethyl 2-furoate
CCOC(=O)c1occc1
616-25-1
1-Penten-3-ol
CCC(O)C=C
61-90-5
L-Leucine
CC(C)CC(N)C(O)=O
61931-81-5
cis-3-Hexenyl lactate
CC\C=C/CCOC(=O)C(C)O
620-02-0
5-Methylfurfural
Cc1oc(C=O)cc1
622-78-6
Benzyl isothiocyanate
#VALUE!
623-37-0
3-Hexanol
CCCC(O)CC
623-42-7
Methyl butyrate
CCCC(=O)OC
624-24-8
Methyl valerate
CCCCC(=O)OC
625-84-3
25-Dimethylpyrrole
CC1=CC=C(C)N1
626-93-7
2-Hexanol
CCCCC(C)O
627-90-7
Ethyl undecanoate
CCCCCCCCCCC(=O)OCC
628-63-7
Pentyl acetate
CCCCCOC(C)=O
629-19-6
Dipropyl disulfide
CCCSSCCC
629-33-4
Hexyl formate
CCCCCCOC=O
63012-97-5
2-Methyl-3-methylthiofuran
CSC1=C(C)OC=C1
6309-51-9
Isoamyl laurate
CCCCCCCCCCCC(=O)OCCC(C)C
63-68-3
L-Methionine
CSCCC(N)C(O)=O
637-64-9
Tetrahydrofurfuryl acetate
CC(=O)OCC1CCCO1
6378-65-0
Hexyl hexanoate
CCCCCCOC(=O)CCCCC
638-25-5
N-Amyl octanoate
CCCCCCCC(=O)OCCCCC
63-91-2
L-Phenylalanine
NC(Cc1ccccc1)C(O)=O
64-04-0
Phenethylamine
NCCc1ccccc1
64-17-5
Ethanol
CCO
64-19-7
Acetic acid
CC(O)=O
64275-73-6
cis-5-Octen-1-ol
CC\C=C/CCCCO
645-56-7
4-Propylphenol
CCCc1ccc(O)cc1
646-07-1
4-Methylvaleric acid
CC(C)CCC(O)=O
6485-40-1
(−)-Carvone
CC(=C)[C@@H]1CC=C(C)C(=O)C1
65405-77-8
cis-3-Hexenyl salicylate
CC/C=C/CCOC(=O)c1ccccc1O
656-53-1
4-Methyl-5-thiazoleethanol acetate
CC(=O)OCCc1scnc1C
65-85-0
Benzoic acid
OC(=O)c1ccccc1
66-25-1
Hexanal
CCCCCC=O
67-03-8
Thiamine hydrochloride
[H+].[Cl−].[Cl−].Cc1ncc(C[n+]2csc(CCO)c2C)c(N)n1
6728-26-3
2-Hexenal
CCC/C=C/C=O
6728-31-0
cis-4-Heptenal
CC/C=C/CCC=O
67-56-1
Methanol
CO
67-63-0
Isopropyl alcohol
CC(C)O
67-64-1
Acetone
CC(C)=O
67-68-5
Methyl sulfoxide
C[S](C)=O
67-71-0
Methyl sulfone
C[S](C)(=O)=O
67715-80-4
2-Methyl-4-propyl-13-oxathiane
CCCC1CCOC(C)S1
67883-79-8
cis-3-Hexenyl tiglate
CC/C=C/CCOC(=O)C(/C)=C\C
6789-88-4
Hexyl benzoate
CCCCCCOC(=O)c1ccccc1
6790-58-5
Ambrox
CC1(C)CCCC2C1CCC3(C)OCCC23
693-54-9
2-Decanone
CCCCCCCCC(C)=O
693-95-8
4-Methylthiazole
Cc1cscn1
698-10-2
5-Ethyl-3-hydroxy-4-methyl-2(5H)-furanone (replicate)
CCC1OC(=O)C(=C1C)O
705-86-2
δ-Decalactone
CCCCCC1CCCC(=O)O1
706-14-9
γ-Decalactone
CCCCCCC1CCC(=O)O1
71-00-1
L-Histidine
O=C(O)C(N)CC1=CNC=N1
710-04-3
δ-Undecalactone
CCCCCCC1CCCC(=O)O1
71-23-8
Propanol
CCCO
71-36-3
Butanol
CCCCO
71-41-0
Pentanol
CCCCCO
71-43-2
Benzene
c1ccccc1
7149-32-8
Phenethyl 2-furoate
O=C(OCCc1ccccc1)c2occc2
7217-59-6
2-Methoxythiophenol
COc1ccccc1S
72-18-4
L-Valine
CC(C)C(N)C(O)=O
7452-79-1
Ethyl 2-methylbutyrate
CCOC(=O)C(C)CC
74-79-3
L-Arginine
NC(CCCN=C(N)N)C(O)=O
7493-63-2
Allyl anthranilate
Nc1ccccc1C(=O)OCC=C
7493-69-8
Allyl 2-ethylbutyrate
CCC(CC)C(=O)OCC=C
7493-71-2
Allyl tiglate
C\C=C(/C)C(=O)OCC=C
75-07-0
Acetaldehyde
CC=O
75-18-3
Methyl sulfide
CSC
7540-51-4
(−)-Citronellol
C[C@H](CCO)CCC=C(C)C
7541-49-3
Phytol
CC(C)CCC[C@@H](C)CCC[C@@H](C)CCC/C(C)=C/CO
75-47-8
Iodoform
IC(I)I
7553-56-2
Iodine
II
7554-12-3
Diethyl malate
CCOC(=O)CC(O)C(=O)OCC
7558-79-4
Sodium phosphate dibasic
[Na+].[Na+].O[P]([O−])([O−])=O
75-65-0
tert-Butanol
CC(C)(C)O
76-22-2
Camphor
CC1(C)C2CCC1(C)C(=O)C2
76-49-3
Bornyl acetate
CC(=O)OC1CC2CCC1(C)C2(C)C
765-70-8
Methylcyclopentenolone
CC1CCC(=O)C1=O
7732-18-5
Water
O
7764-50-3
D-Dihydrocarvone
CC1CCC(CC1=O)C(C)=C
7779-41-1
Decanal dimethyl acetal
CCCCCCCCCC(OC)OC
7779-50-2
Ω-6-Hexadecenlactone
O=C1OCCCCCCCCCC=CCCCC1
7779-65-9
Isoamyl cinnamate
CC(C)CCOC(=O)/C=C/c1ccccc1
77-83-8
Ethyl 3-methyl-3-phenylglycidate
CCOC(=O)C1OC1(C)c2ccccc2
7785-70-8
α-Pinene
CC1=CC[C@@H]2C[C@H]1C2(C)C
7786-58-5
Octyl isovalerate
CCCCCCCCOC(=O)CC(C)C
7787-20-4
L-Fenchone
CC1(C)[C@H]2CC[C@](C)(C2)C1=O
77-90-7
Tributyl-2-Acetylcitrate
CCCCOC(=O)CC(CC(=O)OCCCC)(OC(C)=O)C(=O)OCCCC
77-92-9
Citric acid
OC(=O)CC(O)(CC(O)=O)C(O)=O
77-93-0
Triethyl citrate
CCOC(=O)CC(O)(CC(=O)OCC)C(=O)OCC
78-59-1
Isophorone
CC1=CC(=O)CC(C)(C)C1
78-70-6
Linalool
CC(C)=CCCC(C)(O)C=C
78761-38-3
trans-Cinnamyl propionate
CCC(=O)OC=Cc1ccccc1
78-83-1
2-Methyl-1-propanol
CC(C)CO
78-84-2
Isobutyraldehyde
CC(C)C=O
78-93-3
2-Butanone
CCC(C)=O
79-09-4
Propionic acid
CCC(O)=O
79-20-9
Methyl acetate
COC(C)=O
79-31-2
Isobutyric acid
CC(C)C(O)=O
79-77-6
β-Ionone
CC(=O)/C=C/C1=C(C)CCCC1(C)C
80-62-6
Methyl methacrylate
COC(=O)C(C)=C
81-14-1
Musk ketone
CC(=O)c1c(C)c(c(c(c1C)[N+]([O−])=O)C(C)(C)C)[N+]([O-])=O
81925-81-7
5-Methyl-2-hepten-4-one
CCC(C)C(=O)\C=C\C
821-41-0
5-Hexen-1-ol
OCCCCC=C
821-55-6
2-Nonanone
CCCCCCCC(C)=O
823-22-3
δ-Hexalactone
CC1CCCC(=O)O1
825-51-4
Decahydro-2-naphthol
OC1CCC2CCCCC2C1
85-91-6
Dimethyl anthranilate
CNc1ccccc1C(=O)OC
87-22-9
Phenethyl salicylate
Oc1ccccc1C(=O)OCCc2ccccc2
88-15-3
2-Acetylthiophene
CC(=O)c1sccc1
881-68-5
Vanillin acetate
COc1cc(C=O)ccc1OC(C)=O
882-33-7
Phenyl disulfide
S(Sc1ccccc1)c2ccccc2
88-69-7
2-Isopropylphenol
CC(C)c1ccccc1O
89-79-2
(−)-Isopulegol
CC1CCC(C(O)C1)C(C)=C
89-83-8
Thymol
CC(C)c1ccc(C)cc1O
90-00-6
2-Ethylphenol
CCc1ccccc1O
90-02-8
Salicylaldehyde
Oc1ccccc1C=O
90-05-1
Guaiacol
COc1ccccc1O
91-22-5
Quinoline
c1ccc2ncccc2c1
91-62-3
6-Methyl quinoline
Cc1ccc2ncccc2c1
91-64-5
Coumarin
O=C1Oc2ccccc2C=C1
92-48-8
6-Methylcoumarin
Cc1ccc2OC(=O)C=Cc2c1
92-52-4
Biphenyl
c1ccc(cc1)c2ccccc2
927-49-1
6-Undecanone
CCCCCC(=O)CCCCC
928-96-1
cis-3-Hexen-1-ol
CC\C=C/CCO
93-04-9
Nerolin Yara Yara
COc1ccc2ccccc2c1
93-08-3
Methyl β-naphthyl ketone
CC(=O)c1ccc2ccccc2c1
93-15-2
4-Allyl-12-dimethoxy benzene
COc1ccc(CC=C)cc1OC
93-16-3
Methyl isoeugenol
COc1ccc(\C=C\C)cc1OC
93-28-7
Eugenyl acetate
COc1cc(CC=C)ccc1OC(C)=O
93-29-8
Isoeugenol acetate
COc1cc(/C=C/C)ccc1OC(C)=O
93-51-6
2-Methoxy-4-methylphenol
COc1cc(C)ccc1O
93-54-9
1-Phenyl-1-propanol
CCC(O)c1ccccc1
93-58-3
Methyl benzoate
COC(=O)c1ccccc1
93-60-7
Methyl nicotinate
COC(=O)c1cccnc1
93-89-0
Ethyl benzoate
CCOC(=O)c1ccccc1
93905-03-4
2-Methoxy-3(5 or 6)-isopropylpyrazine
COc1cncc(n1)C(C)C
94278-27-0
Ethyl 3-(furfurylthio)propionate
CCOC(=O)CCSCc1occc1
94-47-3
Phenethyl benzoate
O=C(OCCc1ccccc1)c2ccccc2
94-62-2
Piperine
O=C(/C=C/C=C/c1ccc2OCOc2c1)N3CCCCC3
96-22-0
3-Pentanone
CCC(=O)CC
96-48-0
4-Hydroxybutanoic acid lactone
O=C1CCCO1
97-42-7
Carvyl acetate
CC(=C)C1CC=C(C)C(C1)OC(C)=O
97-53-0
Eugenol
COc1cc(CC=C)ccc1O
97-54-1
Isoeugenol
COc1cc(\C=C\C)ccc1O
97-61-0
2-Methylpentanoic acid
CCCC(C)C(O)=O
97-99-4
Tetrahydrofurfuryl alcohol
OCC1CCCO1
98-00-0
Furfuryl alcohol
OCc1occc1
98-85-1
α-Methylbenzyl alcohol
CC(O)c1ccccc1
98-86-2
Acetophenone
CC(=O)c1ccccc1
99-87-6
para-Cymene
CC(C)c1ccc(C)cc1
Acknowledgments
References